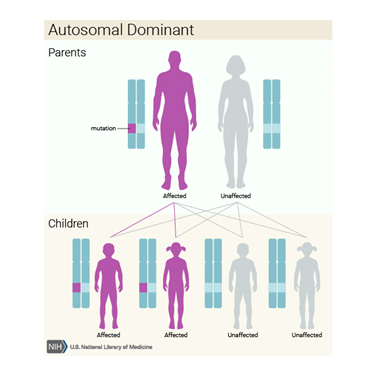
Laband sendromu oldukça nadir
görülen genetik hastalıktır ve aynı zamanda Zimmerman-Laband sendromu olarakta
adlandırılır. Laband sendromu kafa, yüz, eller ve ayaklarda gözlemlenen
anormallikler karakterize edilir. Laband sendromuna sahip çoğu çocukta normal
olmayan büyüklükte diş etleri gözlemlenir ve bu sebeple çiğneme, yutma, veya
konuşma kabiliyetleri etkilenebilir. Ek olarak, Laband sendromlu yeni
doğanlarda oldukça uzun, ince el ve ayak parmakları, eksik ya da şekilsiz
tırnaklar gözlemlenebilir. Bazı rapor edilen vakalarda aynı zamanda zihinsel
gerilikte gözlemlenmiştir. Laband sendromu çoğu vakada otozomal dominant olarak
aktarıldığı bulunmuştur. Fakat, otozomal çekinik olarak aktarıldığı vakalarda
gözlemlenmiştir.
Belirti ve
Semptomlar
Genel bilgide de bahsedildiği üzere
laband sendromu oldukça nadir hastalıktır. Yüz, kafa, eller ve ayaklarda
anormallikler görülür. Laband sendromlu bebeklerde doğuştan itibaren gelen el
ve ayak parmaklarında anormal yapılar gözlemlenebilir. Diğer semptomlar,
ilerleyen çocukluk dönemlerine kadar gözlemlenmeyebilir.
Laband sendromlu çoğu çocukta
gereğinden büyük diş etlerinin varlığı gözlemlenebilir. Diş etlerinin aşırı
büyümesi farklı semptomlara da neden olur. Bunlardan birkaç tanesi ise dişlerin
çıkmasının gecikmesi, dişlerin birbiriyle olması gerektiği gibi yan yana
olmamasından dolayı kusurlu bir diş kapanışı olarak sıralanabilir. Sadece
bunlarla da kalmayıp çiğneme problemleriyle birlikte konuşma sıkıntıları, salya
artışına veya ağızın anormal bir şekilde fazla kurumasına, yutkunmada
zorluklara, ağız kenarlarında yara oluşumuna, dişlerin erken kaybına da sebep
olabilir. Bazı uç vakalarda ise diş etlerinin bütün dişleri kaplayacak kadar
büyümesi gözlemlenmiştir.
Laband sendromuna sahip bireylerde
yukarıda da bahsedilen yüz anormallikleri erken çocukluk döneminde ortaya çıkıp
ergen boyunca anormallikler ağırlaşabilir. Bahsi geçen anormalliklerin içinde
dar bir yüz yapısı, aşırı büyümüş dil, dudaklar, burun, kulaklardan herhangi
bir veya birden fazlası olabilir. Kulaktaki ve burundaki kıkırdak yapıları
Laband sendromlu bireyler normalden daha yumuşak olması gözlemlenebilir ve ek
olarak bazı Laband sendromlu çocuklarda aşırı saç uzaması da gözlemlenmiştir.
Çoğu Laband vakalarında el ve
ayaklarda şekil bozuklukları gözlemlenebilir. Şekil bozuklukları oldukça uzun
ve ince el, ayak parmakları (araknodaktili) ile beraber şişik parmak uçları
içerebilir. Bazı vakalarda ise el, ayak şekil bozukları parmak uçlarının
kusurlu oluşumunu, el ile el parmaklarının eklemlerinin olağandışı bir seviyede
esnek olmasını içermiştir. Ek olarak, el ve ayak tırnaklarında, özellikle baş
parmaklarda, kusurlu oluşum veya oluşmama gözlemlenebilir.
Bazı Laband sendromlu bireylerde
farklı fiziksel semptomlarda gözlemlenmiştir. Bazılarında yukarıda
bahsedilenlere ek olarak iskeletsel ve omurgasal anormallikler, oldukça büyük
karaciğer gözlemlenebilir. Normal zeka seviyesi bazı vakalarda gözlemlenirken
bazılarında ise zeka geriliği gözlemlenmiştir.
Genetik Görülme
Sıklığı
Laband sendromu aşırı nadir genetik bir hastalıktır. Ek
olarak, dişi ve erkekleri etkileme oranı olarak bir farklılık
gözlemlenmemiştir. 1928’de hastalık ilk tanımlandığından beri literatürde 30
farklı vaka bulunmuştur.
Kalıtım
Deseni
Çoğunlukla Laband sendromu otozomal dominant olarak
kalıtıldığı gözlemlenirken azınlıkta olarak otozomal çekinik olarak ta
aktarıldığı rastlanmıştır.
Teşhis
Yöntemleri ve Tedaviler
Laband sendromu ile sonuçlanan çoğu
teşhis erken çocukluk döneminde olduğu fark edilir. Teşhisten özenli bir klinik
inceleme ile ayrıntılı bir hasta geçmişi, özelleştirilmiş testler, el ve ayak
parmaklarının röntgen görüntüsü, burnun, kulakların, dudakların ve dilin
ayrıntılı incelenmesi beraber doğrulama yolu takip edilir. Kesin doğrulanma
yolu olan aşırı büyümüş diş etleri olması süt dişlerinin çıkışına kadar
gözlemlenmeyebilir.
Laband sendromlu bireyin tedavisi
kişide gözlemlenen semptomları tedavi etme yolundadır. Kapsamlı bir tedavi
pediatri uzmanlarını, diş anormalliklerini incelemek ve düzeltmek üzerinde
uzmanlar (dişçiler, ortodonti uzmanları), kemiksel anormallikleri düzelten
uzmanlar (ortopedi uzmanları), diş eti hastalıkları uzmanı (periodontist),
farklı mesleklerden uzmanların beraber çalışmasını gerektirebilir.
Ek olarak bahsetmek gerekir ki bazı
vakalarda doğru dürüst uygulanan ağız hijyeni dişetlerinde oluşan
anormallikleri oluşumunu geçiktirdiği veya azalttığı gözlemlenmiştir.
Dişetindeki anormallikler bazı vakalarda ameliyatsal olarak tedavi edilebildiği
de gözlemlenmiştir. Fakat, bunlara rağmen diş etlerinin aşırı büyümesi hala
görüldüğü vakalarda olmuştur. Diş etlerinin dişleri tamamen kapattığı
durumlarda düzgün bir ağız hijyeni sağlamak hasta için oldukça zorlaşabildiği
gözlemlenmiştir.
Semptomlarda özet geçildiği üzere
Laband sendromlu çocuklarda karaciğer veya dalak büyümesi riski olduğundan
dolayı erken teşhis çocuklar başta olmak üzere oldukça önemlidir.
Pitt-Hopkins
sendromu zihinsel engellilik, gelişimsel gecikme, solunum problemleri, nöbetler
(epilepsi), tipik yüz yapısı ve yüksek miyopi özellikleri ile karakterize olan
genetik bir sendromdur. TCF4 geninin mutasyonu sonucu ortaya çıkar ve otozomal
dominant karakterdedir. İlk defa 1978 yılında David Pitt ve Ian Hopkins
tarafından tanımlanmıştır. Nedeni bilinse de tedavisi için bir ilacı
bulunmamaktadır.
Genetik Değişiklikler/Etken Faktörler
Pitt-Hopkins
sendromuna TCF4 genindeki mutasyon ya da 18. kromozomun TCF4 genini içeren
bölgesindeki delesyon sebep olmaktadır. Otozomal baskın (dominant) olarak
kalıtılır fakat ailesinde Pitt-Hopkins sendromu öyküsü olmadığı halde de
mutasyon sonucu ortaya çıkmaktadır.
TCF4
geni Pitt-Hopkins sendromundan başka Şizofreni, Otizm, Fuchs Kornea Distrofisi
ve Karaciğer hastalığı olmak üzere çeşitli hastalıklarda da rol oynar.
Belirti ve Semptomlar
Pitt-Hopkins
sendromunun birçok belirti ve semptomu bulunmaktadır. Semptomlar ve şiddetleri
kişiden kişiye değişebilir. Erken semptomlar bebek doğduktan sonra bir sene
içinde görülmektedir. Çok düşük kas tonusu (hipotoni) ve gelişim geriliği
görülmektedir. Bazı bebeklerde baş boyutu küçüktür (mikrosefali). Çocuklar
yürümeye beklenenden aylar hatta yıllar sonra başlayabilir. Bazıları ise
bağımsız olarak yürüme yeteneğini kazanamazlar. Konuşma gecikmiştir. Bazıları
birkaç kelime söylemeyi öğrenebilirken çoğu konuşamaz. Bununla birlikte
bazıları basit yönergeleri anlayıp uygulayabilir. Zihinsel engellilik seviyesi
genellikle orta ila şiddetlidir.
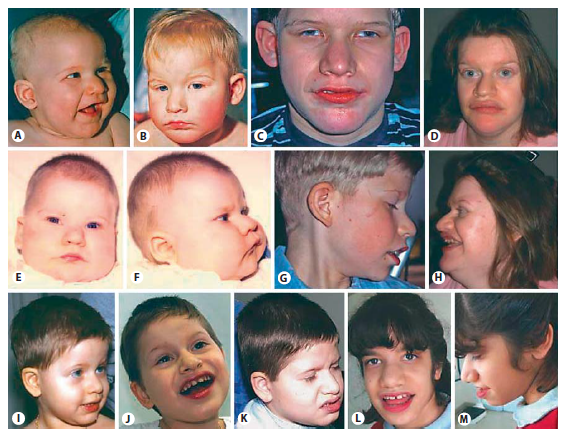
Yüz özellikleri çukur gözler, çıkık burun, aşağı yönelmiş burun ucu, geniş burun delikleri ve burun kemeri, kısa filtrum, geniş ağız, geniş aralıklı dişler ve çıkık çene ile ayırt edilir. Bu özellikler yaş ile daha belirgin hale gelir. (Şekil 1)
Şekil 1. Pitt-Hopkins sendromu hastalarında yüz özellikleri. Hasta 1 A (6 aylık), B (18 aylık) ve C (14 yaş). Hasta 6 D ve H (29 yaş). Hasta 2 E, F (6 aylık) ve G (11 yaş). Hasta 3 I ( 3 yaş), J (6 yaş) ve K (8 yaş). Hasta 4 L ve M (12.5 yaş).
Otizm
spektrum bozukluğu belirtileri görülebilir. Beslenme sırasında alışılmadık davranışlar,
agresif davranışlar, anksiyete, stereotipik el ve kafa hareketleri görülür. Çoğu
çocukta mutlu bir yüz ifadesi vardır. Uykuda kaybolan, soluk alıp verme
problemi görülür. Bunlar genelde anksiyete, heyecanlanma ya da yorulma sonucu
gerçekleşir. Hızlı nefes alıp verme ardından nefes almamaya veya nefes almaya
çalışma davranışları birbirini takip eder. Pitt-Hopkins sendromuna sahip
kişilerin neredeyse yarısında nöbet görülür. Nöbetler çocukluk ya da gençlik
döneminde herhangi bir zamanda başlayabilir. Uyuya kalma ya da uykusuzluk gibi
sorunlar görülebilir. Hastaların yarısından azında gastroözofageal reflu
görülür. Miyopi iki yaşından önce görülebilir ve şiddetli olabilir. Bunun
haricinde şaşılık ve astigmatizm de görülür. İskelet sisteminde skolyoz,
düztabanlık, yumru ayak, küçük el ve ayaklar, kavisli ya da bükük parmaklar,
geniş parmak uçları ya da gittikçe incelen parmaklar görülebildiği rapor
edilmiştir. Ayak parmakları üst üste binmiş halde olabilir. Acı eşikleri
yüksektir. Etkilenen erkeklerin yaklaşık üçte birinde testislerden biri ya da
her ikisi de skrotuma inmemiştir.
Genetik Görülme Sıklığı
Pitt-Hopkins sendromunun oldukça
nadir olduğu düşünülmektedir. Dünya çapında 500 kişinin bu sendroma sahip
olduğu rapor edilmiştir. Kadın ve erkekte görülme sıklığı eşittir.
Kalıtım Paterni/Deseni
Otozomal
baskın (dominant) olarak kalıtılır. Baskın genetik hastalıkların ortaya çıkması
için anormal genden bir tane bulunması yeterlidir. Bu gen anne veya babadan
gelmiş olabilir.
Üreme
hattı mozaikliği görülebilir. Bu durumda anne veya baba TCF4 gen mutasyonunu
vücut hücrelerinde taşımaz yani etkilenmemiştir. Fakat üreme hücreleri (yumurta
ya da sperm) mutasyona sahiptir. Yumurta veya spermlerden biri mutasyona
sahipse hastalık yavru bireyde görülür. Ebeveynlerin kanlarından yapılan testte
mutasyon negatif çıksa bile sonraki çocuklarının da etkilenmiş olma
olasılıkları yaklaşık %1-2 kadardır.
Teşhis Yöntemleri ve Tedaviler
Pitt-Hopkins
sendromunun teşhisi detaylı hasta hikayesi, klinik bulgular ve semptomun
karakteristik özelliklerinin tanımlanmasıyla gerçekleştirilir. Ancak bu sendrom
ve diğer bazı nörolojik bozukluklarda görülen semptomlar benzerlik
göstermektedir. EEG ve beyin MRI görüntülemesiyle ve genetik analizler
sonucunda TCF4 geninde meydana gelmiş mutasyon ya da delesyon tespit edilerek
teşhis desteklenir.
Bilinen
bir tedavisi yoktur. Tedaviler semptomları gidermek için uygulanır.
Hastalıkla İlişkili Genler
TCF4
genindeki mutasyonlar Pitt-Hopkins sendromuna neden olmaktadır. Bu gen 18.kromozomun
uzun kolunda bulunmaktadır (18q21.2). TCF4 geni, vücutta birçok fonksiyonda
kritik rol oynayan proteinlerin yapımı için bilgiler içerir. Bu gende mutasyon
gerçekleştiğinde oluşan protein hatalı ve etkisiz olabilir, eksik veya fazla
üretilebilir. TCF4 geni, transkripsiyon faktörü olarak da bilinen TCF4
proteinini sentezlenmesini sağlar. Bu proteinin farklı gelişim evrelerinde hücre
farklılaşması ve hücre ölümü gerçekleştirmek gibi önemli görevleri vardır.
İnsan gelişiminin erken dönemlerinde sinir sistemi gelişimiyle ilişkili olarak
yüksek düzeyde eksprese edilir.
Hastalığın Diğer İsimleri
Hastalık
PHS ve PTHS kısaltmalarıyla da adlandırılır.
Peippo M., Ignatius J. Pitt-Hopkins Syndrome. Mol Syndromol
2011;2:171–180
Sweatt
J. D. Pitt–Hopkins Syndrome: intellectual disability due to loss of
TCF4-regulated gene transcription. Experimental & Molecular Medicine (2013)
45
Oculodentodigital Displazi, özellikle gözleri (oculo),
dişleri (dento) ve parmakları (digital) etkileyen sistmik bir rahatsızlıktır.
Küçük gözler, görme kaybı, kayıp dişler, tekrarlayan diş çürükleri ve
parmaklardaki fazladan kemiksi büyümeler gibi semptomlar verebilir. GJA1 geninde mevcut bir mutasyon sonucu
görülebilecek bu displazinin otozomal dominant kalıtıldığı düşünülmektedir.
Oculodentodigital Displazi, fizik muayene ile teşhis edilip, genetik testlerle
doğrulanabilir. Hastalığın idare edilmesinde semptomlara yönelik tedaviler
kullanılabilir. Aynı zamanda erken teşhisle hayat kalitesi arttırılabilir.
Genetik
Değişikler/ Etken Faktörler
GJA1
genindeki (6.
Kromozomun uzun kolunda: 6q21-q23.2) mutasyonlar Oculodentodigital
Displaziye yol açmaktadır. GJA1 geni,
Connexin 43 proteini yapımından sorumludur. Bu protein, hücreler arası direkt
iletişimi sağlayan aralıklı bağlantılar için bir alt ünite üretir. Connexin 43
tarafından üretilmiş aralıklı bağlantılar vücudun çeşitli yerlerinde bulunur.
GJA1
genindeki
bir mutasyon, anormal Connexin 43 üretimine sebep olur. Bu anormal proteinle
oluşmuş kanallar genellikle kapalıdır. Bazı mutasyonlar sonucunda, Connexin 43
kanal oluşturması gereken hücre yüzeyine ulaşma yetisini kaybeder. Bu sebeple
hücreler arası iletişim bozulur ve sonucunda hücrelerin büyüme ve özelleşme
süreçlerinde hatalar olur. Oculodentodigital Displazi’de görülen morfolojik
bozuklukların sebebi de bu hatalardır.
Belirti
ve Semptomlar
Özellikle
4. ve 5. parmaklar arası yapışıklık (sindaktil)
İncelmiş,
hassas diş minesi, buna bağlı olarak eksik dişler ve tekrarlayan kaviteler
Ucu
ince, dar burun delikleri ve dış kanatları incelmiş spesifik bir burun
Yavaş
uzayan, kuru saçlar (hipotrikoz)
Araları
oldukça açık, küçük gözler (mikroftalmi)
Glokom
Şaşılık
(Strabismus)
Görme
kaybı
Mikrosefali
Yarık
damak
Çeşitli
nörolojik semptomlar ( Ataksi, kaslarda spastisite, duyma kaybı, idrar veya
gayta inkontinansı, disartri)
Palmoplantar
Keratoderma
Alt
çenede fazla gelişme
Kafatası
kemiklerinde anormal kalınlaşma
Anormal
genişlikte köprücük kemikleri
Kulak
memelerinde anormal kalsiyum birikmeleri
Kardiyak
malformasyonlar
Genetik
Görülme Sıklığı
Oculodentodigital Displazi kadınları ve erkekleri eşit
sıklıkta etkileyen oldukça nadir bir hastalıktır. Medikal literatüre geçmiş 85
vaka bulunmaktadır. Otozomal resesif formu yalnızca 5 vakada saptanmıştır.
İnsidans tam olarak bilinmemektedir. Çoğu vakanın teşhissiz kaldığı düşünülmektedir.
Kalıtım
Paterni
Oculodentodigital Displazi, çoğunlukla otozomal
dominant olarak kalıtılmaktadır. Ancak literatürde aile öyküsü olmadan yeni
mutasyonlarla oluşmuş bireyler de mevcuttur.
Teşhis
Yöntemleri/ Tedaviler
Teşhis, fizik muayene ve genetik testlerle mümkündür.
Tedavi ise semptomlara yöneliktir.
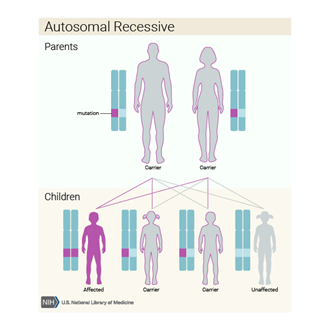
N-Acetylglutamate
Sentaz (NAGS) Eksiliği otozomal çekinik gen ile taşınan, hiperamonyemiye neden
olan üre siklüs bozunluklarındandır. Bu hastalık hem vücudun proteinleri
işlemesini hem de vücuttan amonyak atılımını etkileyen metabolik bir
bozukluktur. Hastalığın sonucu olarak kişi vücudundan amonyağı atamaz ve bunun
sonucu olarak vücutta fazlaca amonyak birikir. Amonyak, vücudun proteinleri
işlemesiyle ortaya çıkar ve vücutta çokça birikirse kişi için zehirli hale
gelir.
Epidemiyoloji
Çok
nadir ve tam olarak yaygınlığı bilinmemektedir. Ancak dünya genelinde 1-2
milyon insanın bu hastalığa sahip olduğu tahmin edilmektedir.
Klinik Bilgi
Hastalık her yaşta ortaya
çıkabilmektedir, ancak çoğunlukla yenidoğanlarda görülmektedir. Klinik görünümü
değişiklik göstermekle beraber en yaygın olanları; kusma, hiperaktivite,
letarji (uyuşukluk), diyare (ishal), beslenmede isteksizlik, nöbet, hipotoni,
geç psikomotor gelişimi ve solunum bozukluklarıdır. Hiperamonyemi sonuçları ağır olan bir hastalıktır ve çoğunlukla
hiperamonemik komaya neden olmaktadır.
Etiyoloji
Birincil rahatsızlık NAGS
geninde (17q21.31) oluşan mutasyonlar sonucu ortaya çıkmaktadır ve kısmı NAGS
aktivitesi eksikliğine neden olmaktadır. NAGS’ın ürünü NAG (N-acetylglutamate),
üreojenezin birinci basamağını katalize eden enzim olan korbomil fosfat sentez I’in
(CPSI) allosterik aktivatörüdür. N-Acetylglutamate
Sentaz Eksiliği, organik asitik hastalıklar, yağ asidi metabolizmasında oluşan
bozukluklar veya valproik asit tedavisi sonucunda da oluşabilmektedir.
Genetik Yatkınlık
N-Acetylglutamate
Sentaz Eksiliği’ne, 17q21 kromozomunda bulunan N-acetylglutamate sentaz geninin
mutasyona uğraması neden olmaktadır. Bu hastalık nesilden nesile otozomal
çekinik gen ile aktarılmaktadır. Bu yüzden kişinin bu hastalığa sahip olabilmesi için her
hücredeki sorumlu geninin ikisinin de mutasyona uğramış olması gerekmektedir.
Hasta bireyler, mutasyona uğramış bir geni anneden bir diğer geni de babadan
almaktadır. Bu durumda ebeveynler taşıyıcı olarak adlandırılmaktadır.
Taşıyıcılar otozomal çekinik gene sahip oldukları için bu hastalığın belirti
veya semptomlarını göstermezler. İki otozomal çekinik gene sahip bireyin çocuğu
olduğunda, bu çocuk %25 ihtimalle hasta, %50 ihtimalle taşıyıcı ve %25
ihtimalle ne hasta ne de taşıyıcı olacaktır.
Belirti ve Semptomları
N-Acetylglutamate
Sentaz Eksiliği’ne sahip hastaların %30-%79’unda çocuklukta görülen kas
hipotonisi, mide bulantısı, kusmaya rastlanmakta olup; %5-%29’unda da akut
hiperamonyemi ve gergin ruh haline rastlanmaktadır. Ayrıca hasta bireylerde
koma durumu, öğrenme güçlüğü, zihin bulanıklığı, kordinasyon eksikliği ve gelişimsel
gecikme görülebilmektedir. Bu semptomlar kişiden kişiye göre farklılık
göstermektedir ve kişi bütün semptomlara sahip olmak zorunda değildir. Semptomları
ve belirtileri ileri yaşlarda göstermeye başlayan insanlar, erken yaşlarda
gösterenlere veya besin tüketiminde proteine fazlaca yer veren insanlara göre
belirti ve semptomları daha hafif yaşarlar. Yenidoğanlarda N-Acetylglutamate
Sentaz Eksiliği’nin semptomları enerji eksikliği, yemek yemede isteksizlik,
nöbet, anormal vücut hareketleri, çok düzensiz ve zor nefes alma, değişken vücut
ısısıdır.
Teşhis Konulması
Nadir
görülen bir genetik hastalığın tanısının konması zor olduğu için doktorlar
genellikle kişinin tıbbi geçmişini, şüphe duyulan semptomlarını incelemekte;
fiziksel muayne ve laboratuvar testi sonuçlarının değerlendirilmesiyle de hastalık
teşhisini koymaktadır. Ayrıca hastalık karaciğerde azalış gösteren NAGS
aktivitesinden de anlaşılabileceği için teşhis DNA analizi ile de desteklenebilmektedir.
Hastalığın ana ayırıcı tanısı karbomil fosfat sentezi eksikliğidir.
Tedavi Edilmesi
N-Acetylglutamate
Sentaz Eksiliği hastaları için uygulanan ana tedavi günlük dozlarla karglumik
asit almaktır. Karglumik asit yapısal bakımdan NAGS’a benzemektedir ve korbomil fosfat sentez I’i (CPSI) aktive etmektedir.
Diğer
İsimleri
N-Acetylglutamate
Sentaz Eksiliği’ne Bağlı Hiperamonyemi
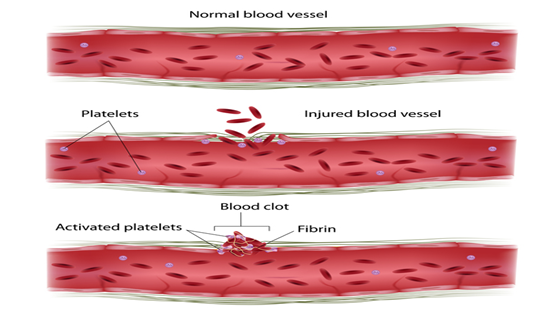
Faktör XI eksikliği ,
kanın pıhtılaşmasında rol alan faktör XI proteininin bir kıtlığına
(eksikliğine) bağlı olarak anormal kanamaya neden olabilecek bir hastalıktır. Bu durum, faktör XI proteininin eksiklik derecesine
bağlı olarak kısmi veya şiddetli olarak sınıflandırılır. Bununla birlikte,
protein eksikliğinin ciddiyetine bakılmaksızın, çoğu etkilenen birey göreceli
olarak hafif kanama problemlerine sahiptir.Faktör XI sendromuna sahip
kişilerdeki belirtiler aynı aile içerisinde bile değişebilir.Diğer genetik ve
çevresel faktörler hastalığın derecesinin belirlenmesinde etkilidir.
Belirti ve Semptomlar
Faktör XI eksikliği vakalarının
çoğu , faktör XI proteini yapmak için talimatlar veren F11 genindeki mutasyonlardan
kaynaklanır . Bu protein, kan pıhtılarını oluşturan
bir dizi kimyasal reaksiyon olan pıhtılaşma kaskadında rol oynar.Yaralanmaya
yanıt olarak. Bir yaralanmadan sonra pıhtılar kanamayı durdurmak ve kan
damarı onarımını tetiklemek için kan damarlarını kapatır.
F11 genindeki mutasyonlar ,
fonksiyonel faktör XI’nin eksikliğine (eksikliğine) neden olur. Bu
eksiklik pıhtılaşma kademesini bozar, kanın pıhtılaşma sürecini yavaşlatır ve
bu hastalıkla ilişkili kanama sorunlarına yol açar. Kalan fonksiyonel
faktör XI miktarı, özel mutasyona ve F11 geninin bir
veya her iki kopyasının, her hücrede mutasyona sahip olup olmamasına
bağlı olarak değişir . Bununla birlikte, etkilenen bireylerde kanama
problemlerinin ciddiyeti mutlaka kan dolaşımındaki faktör XI miktarına karşılık
gelmez ve aynı aile içinde bile
değişebilir. Diğer genetik ve çevresel faktörler muhtemelen bu
durumun ciddiyetinin belirlenmesinde rol oynar.
Faktör XI eksikliğinin en
sık görülen özelliği , özellikle ağız ve burun içini (travma veya burun boşlukları dahil) travma
veya ameliyat sonrası uzun süreli kanamadır.) veya idrar yolu. Kanama ameliyattan sonra tedavi
edilmezse, cerrahi alanda konjuge kandan (hematom) oluşan katı şişlikler
gelişebilir.
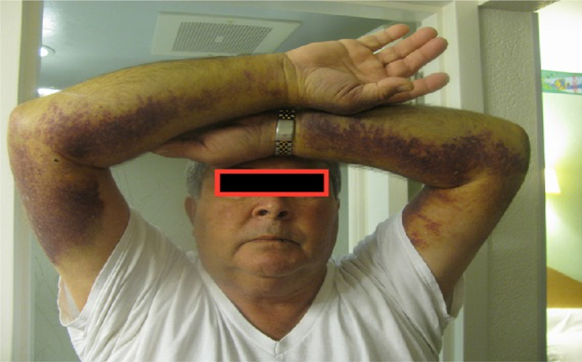
Bu hastalığın diğer belirti ve semptomları sık burun
kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını
içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet
kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip
olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri),
gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın
olmayan faktör XI eksikliği onlar
ağır etkilenen bireylerde oluşabilir rağmen. Diğer kanama bozukluklarında
uzun süreli sakatlığa neden olabilecek kaslara veya eklemlere kanama genellikle
bu durumda meydana gelmez.
Bu hastalığın diğer belirti ve semptomları sık burun
kanaması, kolay morarma, deri altında kanama ve diş etlerinin kanamasını
içerebilir. Bu bozukluğu olan kadınlar ağır veya uzun süreli adet
kanamasına (menoraji) veya doğumdan sonra uzun süreli kanamaya sahip
olabilir. Diğer bazı kanama bozukluklarında aksine, idrar (hematüri),
gastrointestinal sistem veya kafatası boşluğuna spontan kanama yaygın
olmayan faktör XI eksikliği onlar
ağır etkilenen bireylerde oluşabilir
rağmen. Diğer kanama bozukluklarında uzun süreli sakatlığa neden
olabilecek kaslara veya eklemlere kanama genellikle bu durumda meydana gelmez.
Aşağıda bu hastalığı olan kişilerin sahip olabileceği
belirtileri listeler. Çoğu hastalık için semptomlar kişiden kişiye
değişecektir. Aynı hastalığı olan insanlar listelenen tüm belirtilere
sahip olmayabilir. Bu tablo düzenli olarak İnsan Fenotip Ontoloji
((Human Phenotype Ontology [HPO]) isimli veri tabanı tarafından
güncellenmektedir. ([https://hpo.jax.org/)
Faktör
XI eksikliğinin dünya genelinde yaklaşık 1 milyon kişiden
1’ini etkilediği tahmin edilmektedir. Orta ve doğu Avrupa (Aşkenazi)
Yahudi soyuna sahip kişilerde, bu popülasyondaki 450 kişiden 1’inde meydana
gelen şiddetli bozukluk bozukluğu çok daha yaygındır. Araştırmacılar, faktör XI eksikliğinin gerçek
prevalansının bildirilenden daha yüksek olabileceğini öne
sürüyorlar , çünkü hastalığın hafif vakaları çoğu zaman tıbbi yardıma
gelmiyor.
Kalıtım Paterni /Deseni
Otozomal
resesif paternde ciddi faktör XI eksikliği azaldıbu, her bir hücrede F11 geninin
her iki kopyasının da mutasyonlara sahip olduğu anlamına
gelir . Bu bireylerin ebeveynlerinin her biri, mutasyona uğramış
genin bir kopyasını taşır ve kısmi faktör XI
eksikliğine sahiptir ; nadiren durumun ciddi belirtileri ve
semptomlarını gösterirler.
Bazı ailelerde bu durum otozomal dominant paternde kalıtsaldırbu, her
hücrede değiştirilmiş F11 geninin bir
kopyasının , bozukluğa neden olmak için yeterli olduğu anlamına
gelir . Bu gibi durumlarda, etkilenen bir kişinin durumu olan bir
ebeveyni vardır.
Kazanılan faktör XI eksikliği formu kalıtsal
değildir ve ailelerde çalışmaz.
Faktör XI eksikliği
sendromunun tanısı için 3 önemli belirti şu şekildedir:
1)Ağız ve burun içi travma veya ameliyat sonrası uzun
süreli kanama.
2)Sık sık burun kanaması, kolay morarma, deri altında
kanama ve diş etlerinin kanaması.
3)Bu hastalığa sahip olan kadınlarda ağır veya uzun
süreli adet kanaması (menoraji) ve doğumdan sonra uzun süreli kanama.
Ayrıca faktör XI eksikliği sendromu için bazı testler
yapılmaktadır. Ancak hastalık tanısı için gerekli değildir.
1)Genetik Test
Genetik test, kromozom, gen veya proteinlerdeki değişiklikleri tanımlayan
bir tıbbi test türüdür. Genetik testin sonuçları, şüpheli bir genetik
durumu onaylayabilir veya ekarte edebilir veya bir kişinin genetik hastalık
geliştirme veya geçme şansını belirlemeye yardımcı olabilir. Şu anda
1000’den fazla genetik test kullanılıyor ve daha fazlası geliştiriliyor.
Genetik test için çeşitli yöntemler kullanılabilir:
Moleküler genetik testler (veya
gen testleri), genetik bir bozukluğa yol açan varyasyonları veya
mutasyonları tanımlamak için tekli genleri veya kısa DNA uzunluklarını
inceler.
Kromozomal genetik testler,
genetik bir duruma neden olan bir kromozomun ekstra bir kopyası gibi büyük
genetik değişikliklerin olup olmadığını görmek için bütün kromozomları
veya uzun DNA uzunluklarını analiz eder.
Biyokimyasal genetik testler,
proteinlerin miktarını veya aktivite seviyesini inceler; herhangi
birindeki anormallikler, genetik bir bozuklukla sonuçlanan DNA’daki
değişiklikleri gösterebilir.
Genetik test isteğe bağlıdır. Testin sınırlamalar ve risklerin yanı
sıra faydaları olduğu için, test edilip edilmeyeceği konusundaki karar kişisel
ve karmaşık bir karardır. Bir genetikçi veya genetik danışman, testin
artıları ve eksileri hakkında bilgi vererek ve testin sosyal ve duygusal
yönlerini tartışarak yardımcı olabilir.
2)Kısmi Tromboplastin Zamanı(PTT):
Kısmi tromboplastin zamanı (PTT) kanın pıhtılaşmasının ne
kadar sürdüğünü gösteren bir kan testidir. Kanama probleminiz olup
olmadığını veya kanınızın pıhtılaşmaması durumunda size yardımcı olabilir.
3)Protrombin Zamanı(PT):
İlgili bir kan testidir.
Hastalığın Diğer İsimleri
F11 eksikliği
faktör 11 eksikliği
hemofili C
hemofili
C
plazma
tromboplastin öncül eksikliği
PTA
eksikliği
Rosenthal
faktörü eksikliği
Rosenthal
sendromu
Rosenthal
hastalığı
Faktör XI Eksikliği İçin Yurt Dışındaki Kuruluşlar
Koni distrofisi, retinanın koni hücrelerini etkileyen bir grup nadir göz hastalığını tanımlamak için kullanılan genel bir terimdir. Koni distrofisi tanımı, kalıtımla ilgili olan kon disfonksiyonlarını ifade eder. Koni işlev bozukluğu kalıtsal nitelikte olan bir grup fotoreseptör distrofilerinden oluşmaktadır. Retina distrofileri geri dönüşümü olmayan fotoreseptör hasarı ile giden, kalıtsal ve genetiği karmaşık bir grup hastalıktan oluşmaktadır.Koni distrofisi değişken bir şekilde ileriye bakarken azalan görsel netliği (keskinliği), renkleri görme kabiliyetini ve ışığa karşı duyarlılığı (fotofobi) içeren çeşitli semptomlara neden olabilir.
Koni distrofisi iki geniş gruba ayrılabilir – durağan ve
ilerici. Sabit konide distrofi semptomları sabit kalma
eğilimindedir ve genellikle doğumda veya erken çocuklukta görülür. İlerici konide distrofi semptomları yavaş yavaş
zamanla kötüleşir. Koni
distrofisinin birkaç farklı şekli vardır. Koni distrofisinin başlangıç yaşı, progresyonu ve şiddeti, aynı koni
distrofisine sahip kişiler arasında bile bir kişiden diğerine büyük
farklılıklar gösterebilir. Bazı koni
distrofi formları kalıtsaldır; diğer
formların görünürde bir neden olmadan (sporadik olarak) kendiliğinden ortaya
çıktığı görülmektedir.
Retinanın özellikle makulanın, son derece karmaşık bir yapısı
vardır ve fonksiyonları çok sayıda gen tarafından kodlanmaktadır. Herediter
retina distrofilerinde (HRD) genlerdeki çeşitli bozukluklara göre farklı
tutulumlar görülebilmekte ve aynı ailenin farklı bireylerinde değişik klinik
tabloların ortaya çıkabilmesine neden olabilmektedir. Koni distrofisinin çeşitli
formlarını tarif etmek için çeşitli ve kafa karıştırıcı isimler kullanılmıştır.
Koni distrofileri grubu içerisinde aşağıdaki hastalıklar yer almaktadır.
Koni reseptörlerinin konjenital yokluğu:
Rod monokromatizmi (Otozomal Resesif-OR)
Koni monokromatizmi (X’e bağlı resesif-X-LR, OR
Konjenital olmayan progresif kon
distrofileri
Herediter (OR, Otozomal dominant-OD, X-LR)
Kalıtım Özellikleri
Birçok koni distrofi vakası, tanımlanabilir bir neden olmadan (sporadik
olarak) rastgele ortaya çıkar. Bazı formlar, otozomal dominant, otozomal
resesif veya X’e bağlı resesif bir özellik olarak kalıtılır. Kalıtsal koni
distrofi formları koni distrofisine bağlı olan birkaç farklı genden birindeki
mutasyonlardan kaynaklanmaktadır. Bu genler, belirli proteinleri,
özellikle de koni hücrelerinin gelişiminde, fonksiyonunda veya genel sağlığında
yaşamsal rol oynayan proteinleri yapmak için talimatlar içerir.Koni
distrofisine neden olan kesin mekanizmalar tam olarak anlaşılmamıştır.
Koni distrofisinde mutasyona uğramış genler, otozomal dominant, otozomal
resesif veya X’e bağlı resesif özellik olarak kalıtsal olabilir. Genetik
hastalıklar, baba ve anneden alınan kromozomlar üzerindeki belirli bir özellik
için genlerin kombinasyonu ile belirlenir.
Hakim genetik bozukluklar, hastalığın ortaya çıkması için anormal bir
genin sadece bir kopyasının gerekli olduğu durumlarda ortaya
çıkar. Anormal gen, her iki ebeveynden kalıtsal olabilir veya etkilenen
bireyde yeni bir mutasyonun (gen değişimi) sonucu olabilir. Anormal genin
etkilenen ebeveynden yavrulara geçme riski, ortaya çıkan çocuğun cinsiyeti ne olursa
olsun her hamilelik için yüzde 50’dir.
X’e bağlı resesif genetik bozukluklar, X kromozomunda anormal bir genin
neden olduğu durumlardır. Dişilerde iki X kromozomu vardır, ancak X
kromozomlarından biri “kapanır” ve bu kromozomdaki tüm genler inaktive
olur. X kromozomlarından birinde bir hastalık geni bulunan dişiler bu
hastalığın taşıyıcılarıdır. Taşıyıcı dişiler genellikle bozukluğun
semptomlarını göstermezler çünkü genellikle “kapalı” olan anormal genli X
kromozomudur. Bir erkekte bir X kromozomu vardır ve eğer bir hastalık geni
içeren bir X kromozomunu miras alırsa, hastalığı geliştirir. X’e bağlı
bozuklukları olan erkekler, hastalık genini taşıyıcı olacak tüm kızlarına
geçirir. Bir erkek, X’e bağlı bir geni oğullarına geçiremez, çünkü
erkekler X kromozomu yerine Y kromozomunu erkek yavrulara her zaman
geçirir. X’e bağlı bir hastalığın dişi taşıyıcıları, her hamilelikte
kendileri gibi taşıyıcı bir kıza sahip olma şansı, yüzde 25’i taşıyıcı olmayan
bir kıza sahip olma şansı, yüzde 25’i hastalıktan etkilenen bir çocuğa sahip
olma şansı ve Etkilenmemiş bir oğlun olması için yüzde 25 şans.
Daha az sıklıkla, koni distrofisi otozomal resesif bir özellik olarak
kalıtsal olabilir. Resesif genetik bozukluklar, bir birey her bir ebeveynden
aynı özellik için aynı anormal geni aldığında ortaya çıkar. Bir
birey hastalık için bir normal gen ve bir gen alırsa, kişi hastalık için
taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem kusurlu geni geçmesi hem de
etkilenen bir çocuğa sahip olma riski her hamilelikte yüzde 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski
her hamilelikte yüzde 50’dir. Bir
çocuğun her iki ebeveynden normal gen alma ve bu özellik için genetik olarak
normal olma şansı yüzde 25’tir. Risk
erkeklerde ve kadınlarda aynıdır.
Nonsendromik CRD’ler genetik olarak heterojendir (28 gen tanımlanmıştır). En yaygın dört mutasyona uğramış dört gen, otozomal resesif CRD’lerin% 30’u ila 60’ından sorumlu olanABCA4 (1p22.1) ve bildirilen birçok otozomal dominant CRD vakasından sorumlu olan GUCY2D (17p13.1) ve CRX (19q13.33) ve X’e bağlı CRD’lerden sorumlu olan ve RPGR (Xp11.4)’dir
Kalıtım: Otozomal
dominant, Otozomal resesif, X’e bağlı resesif
Başlangıç
Yaşı: Ergenlik, yetişkinlik, çocukluk
Görülme Sıklığı
Konik distrofi, erkekleri ve dişileri, sporadik olarak ortaya çıktığında
veya otozomal dominant veya resesif bir özellik olarak kalıtsal olarak eşit
sayıda etkiler. X’e bağlı resesif konik distrofi formu sadece erkekleri
tamamen etkiler, ancak bazı dişilerde hastalığın hafif belirtileri olabilir. Koni
distrofisinin kesin görülme sıklığı bilinmemektedir ve tahminler tıbbi
literatürde farklılık gösterme eğilimindedir.Çoğu kaynak, insidansı genel
popülasyondaki 30.000 kişide yaklaşık 1 olarak tahmin etmektedir. Koni
distrofisi genellikle erken bebeklik döneminde veya çocukluk döneminde veya
erken yetişkinlikte bulunur. Ancak, bozukluğun yaşlı yetişkinler de dahil
olmak üzere her yaştaki bireylerde geliştiği bildirilmiştir.
Görme prognozu değişkendir, çoğu durumda 40 yaşından önce körlüğe yol
açan erken merkezi görme kaybı ve progresif görsel işlev bozukluğu vardır.
Prevalans: 1-9
/ 100 000
Tanı ve Tedavi
Konik distrofi tanısı, karakteristik semptomların
tanımlanması, ayrıntılı hasta öyküsü, ayrıntılı klinik değerlendirme ve tanı
testi standart bir elektroretinogramdır. Görme keskinliğini, rengi algılama yeteneğini ve bir kişinin görüş
alanını ölçen normal oftalmolojik muayeneler tanı konmasına yardımcı olmak için
kullanılır.
Ayırıcı tanı, diğer kalıtsal
koni bozukluklarını (akromatopsi ve akraba koni disfonksiyon sendromları, koni
distrofisi ve Stargardt hastalığı dahil) ve aynı zamanda fotoreseptör
tutulumunun sekansı (koni fotoreseptörleri çubuk fotoreseptörleri
tarafından takip edilir) ile ayırt edilen çubuk-koni distrofisini, ayrıca
asretinit pigmentoza olarak bilinir içerir.
Bir ERG sırasında, göze özel bir kontakt lens elektrotu
yerleştirmeden önce gözü uyuşturmak için göz damlaları kullanılır. Hasta daha sonra retinayı uyarmak için yanıp sönen ışıkları izler. Doktorlar daha sonra koni ve çubuk hücreler tarafından
üretilen elektrik sinyallerini ölçebilir. Zayıf veya zayıf bir koni hücresi sinyali, koni distrofisini gösterir. ERG testi iki kez yapılır – bir kez aydınlık bir odada
ve bir kere karanlık bir odada. Test
koni ve çubuk hücrelerinin düzgün çalışıp çalışmadığını belirleyebilir.
Koni distrofisinin tedavisi yoktur. Tedavi, her bir bireyde
belirgin olan spesifik semptomlara yöneliktir. Tedavi, aydınlık ortamlarda
renkli merceklerin veya karanlık güneş gözlüklerinin kullanılmasını ve okuma ve
benzeri etkinliklerde yardımcı olması için büyüteçlerin kullanılmasını
içerebilir.
Genetik danışma, etkilenen bireyler ve aileleri için faydalı
olabilir. Diğer tedavi semptomatik ve destekleyicidir.
Referanslar
DERS KİTAPLARI
Heckenlively, John R. Koni Distrofileri ve Dejenerasyonları. In:
Vizyonun Klinik Elektrofizyolojisi, JR Heckenlively ve GB Arden editörlerinin
İlkeleri ve Uygulaması.2006 MIT Press, Cambridge, MA. pp. 795-802
Ho AC, Kahverengi GC, McNamara JA. Ed. Retina: Renk Atlası ve
Klinik Oftalmolojinin Konusu. McGraw-Hill Şirketler, Inc Columbus,
OH. 2003: 146-147.
Tasman W, Jaeger EA, ed. Klinik Oftalmoloji Atlası, 2.
baskı. Lippincott, Williams ve Wilkins. Philadelphia; 2001: 173-174.
Kohl S. Kalıtsal koni ve koni-çubuk distrofilerinin genetik
nedenleri.Ophthalmologe. 2009; 106: 109-115.
Holopigian K, Greenstein VC, Seiple W, Hood DC, Carr RE. Konik
distrofisi olan hastalarda, çubuk ve koni fotoreseptör fonksiyonu. Yatırım
Oftalmol Vis Sci. 2004; 45: 275-281.
Michaelides M, Aligianis IR, Ainsworth JR, vd. CNGB3’te mutasyonla
ilişkili progresif koni distrofisi. Yatırım Oftalmol Vis Sci. 2004;
45: 1975-1982.
Michaelides M, Hunt DM, Moore AT. Koni fonksiyon bozukluğu
sendromları. Br J Oftalmol. 2004; 88: 291-297.
Simunovic milletvekili, Moore AT. Koni distrofileri. Göz
(Lond). 1998; 12: 553-565.
İNTERNETTEN
Openshaw A, Branham K, Heckenlively J. Koni Distrofisini Anlamak. Michigan
üniversitesi. Kellogg Göz Merkezi. Şubat 2008. Erişim tarihi:
http://www.kellogg.umich.edu/patientcare/conditions/Understand-Cone-Dystrophy.pdf
Erişim tarihi: 20 Aralık 2009.
Eş Acquired Hemophilia Hastalıkları: • acquired hemophilia A (AHA) (Klasik hemofili) • acquired hemophilia B (AHB) (Noel hastalığı)
Genel Bilgi Kazanılmış (edinilmiş) hemofili (AH) kanama ile karakterize nadir görülen otoimmün bir hastalıktır. Otoimmün bozukluklar, vücudun bağışıklık sistemi yanlışlıkla sağlıklı hücrelere veya dokulara saldırdığında ortaya çıkar. AH’de vücut, en sık faktör VIII olan pıhtılaşma faktörlerine saldıran antikorlar (inhibitörler olarak bilinir) üretir. Pıhtılaşma faktörleri, kanın normal olarak pıhtılaşması için gereken özel proteinlerdir. Sonuç olarak, etkilenen bireyler, kaslara, cilde ve yumuşak dokuya, ameliyat sırasında veya travma sonrası anormal, kontrolsüz kanama ile ilişkili komplikasyonlar geliştirir. Spesifik semptomlar arasında burun kanaması (burun kanaması), vücutta morluklar, konjekte olmuş kanın (hematomlar) katı şişmesi, idrardaki kan (hematüri) ve gastrointestinal veya ürogenital kanamalar bulunur. AH, ağır vakalarda ciddi, hayatı tehdit edici kanama komplikasyonlarına neden olabilir. Hastaların yaklaşık% 50’sinde, tanımlanabilir bir altta yatan klinik durum vardır; diğer% 50’sinde ise hiçbir neden bilinmemektedir. AH, bazı pıhtılaşma faktörlerinin konjenital eksikliğinden kaynaklanan nadir bir genetik hastalık grubu olan konjenital hemofili’den farklıdır. Ana hemofili şekli X’i bağlayan bir hastalık olan hemofili A’dır (klasik hemofili), erkekleri de kadınları da etkileyebilir.
Faktörü VIII olmayan veya azalmış olan hastalarda konjenital hemofili A (“klasik” hemofili) ve IX olan veya olmayan faktörde IX olanlar konjenital hemofili B (Noel Hastalığı) olarak bilinir.
Genetik Değişiklikler / Etken Faktörler
Nedenleri AH, otoimmün bir hastalıktır. Bağışıklık sistemi yanlışlıkla sağlıklı dokuya, özellikle de pıhtılaşma faktörü olarak bilinen özelleşmiş proteinlere, çoğunlukla pıhtılaşma faktörü VIII’e saldıran antikorlar ürettiğinde ortaya çıkar.
Bağışıklık sistemi normal olarak antikor adı verilen özel proteinler üreterek yabancı bir maddeye tepki verir. Antikorlar, yabancı maddeleri doğrudan yok ederek veya beyaz kan hücrelerinin yok etmesi için onları işaretleyen bir maddeyle kaplayarak çalışır. Antikorlar sağlıklı dokuları hedeflediklerinde, otoantikorlar olarak adlandırılabilirler. Araştırmacılar, tetikleyici bir olayın (bir enfeksiyon veya altta yatan bir bozukluk gibi) bağışıklık sistemini otoantikorlar üretmeye neden olabileceğine inanmaktadır. AH’deki otoantikorlar, inhibitör olarak adlandırılır çünkü etkilenen pıhtılaşma faktörünün fonksiyonunu inhibe ederler.
AH ağırlıklı olarak yaşlıların bir hastalığıdır. Hastaların yaklaşık% 50’sinde, altta yatan bir bozukluk veya tetikleyici olay tanımlanamaz (idiyopatik form). Kalan% 50, lupus, romatoid artrit, multipl skleroz, Sjögren sendromu ve temporal arterit gibi otoimmün rahatsızlıklar dahil olmak üzere birlikte varolan rahatsızlık veya rahatsızlıklara sahiptir; inflamatuar bağırsak hastalığı veya ülseratif kolit; enfeksiyonlar; şeker hastalığı; hepatit; solunum veya dermatolojik hastalıklar; kan (hematolojik) kanser veya bazı katı tümörler veya penisilin veya interferon gibi ilaçlarla ilişkisi. Gebelikle ilişki, esas olarak doğum sonrası dönemde bildirilmektedir.
Belirti vr Semptomlar Hastaların yaklaşık 1 / 3’ünde kanamaları kontrol etmek için tedavi gerekmese de kanama şiddeti değişkendir ve hastaların 1 / 3’ünden fazlasında çoklu kanama atakları vardı. Deri altı kanaması (ekimozlar) AH’nin en sık görülen belirtisidir, bunu kas kanaması (hematom), gastrointestinal (melena), genitoüriner (hematüri) ve retroperitoneal izler. İntrakraniyal kanama nadirdir, ancak ölümcül olabilir. Konjenital hemofili A’nın aksine eklem kanaması nadirdir.
Kanama genellikle sebepsiz olarak oluşur (kendiliğinden). Kanama bölümleri genellikle ağırdır ve hayatı tehdit edici olabilir. Bazı hastalarda, gecikmiş tanı ve ek tıbbi sorunların varlığı genellikle genel şiddetine etki eden faktörlerdir. Yumuşak dokulara kanama hızlı bir şekilde ilerleyebilir, potansiyel olarak kaslar, sinirler ve Bu yapıların sıkışması nedeniyle en sık olarak kol ve bacaklarda bulunan kan damarları etkilenir. Etkilenen Bireyler ayrıca, ameliyat sırasında veya travma sonrası, hatta önemsiz olsa bile, aşırı kanama riski altındadır. Gebe kadınlarda genital ağır kanama, özellikle doğum sonrası (doğum sonrası dönem) ortaya çıkabilir.
Genetik Görülme Sıklığı AH, önceden eşit kanama bozukluğu öyküsü olmayan ve yaklaşık olarak eşit sayıda erkek ve kadın etkilenen bireylerde gelişir. Amerika Birleşik Devletleri’nde, bozukluğun genel popülasyonda yılda 1.000.000 / yılda yaklaşık 0.2-1 kişiyi etkilediği tahmin edilmektedir. Birleşik Krallık’ta, hastalığın 1.000.000 / yılda 1.4’ü etkilediği tahmin edilmektedir. Ancak, etkilenen bireyler teşhis edilmeyebilir veya yanlış teşhis edilebilir, bu da hastalığın genel popülasyondaki gerçek sıklığını belirlemeyi zorlaştırır. Vakaların çoğu faktör VIII (AHA) eksikliğini içerir. Faktör IX (AHB) eksikliğini içeren vaka daha az sayıda tanımlanmıştır.
Kalıtım Paterni/Deseni Hemofili vakalarının çoğunluğu genetiktir, erkekleri etkiler ve çocukluk çağında oldukça erken teşhis edilir. Ancak, daha sonra yaşamda gelişen ve “kazanılmış hemofili” olarak adlandırılan nadir görülen hemofili vakaları vardır.
Edinilmiş hemofili, hastaların milyonda birini etkileyen çok nadir görülen bir durumdur. Genellikle hastalara uzun süreli kanama olduğunda veya kaza v.s. sebeplerle travma sonrası, bacaklarda ve kollarda olağan yaralanma ile ilgili morarmalardan çok farklı görünen geniş morarmalarda teşhisi konur. Basit bir kan testi, pıhtılaşma faktörü VIII eksikliği olup olmadığını belirleyebilir. Eğer tedavi edilmezse, alınan hemofili çok ciddi ve yaşamı tehdit edici olabilir. Bununla birlikte, immünosüpresif ilaçlar ve pıhtılaşma faktörü enjeksiyonları ile teşhis konduktan sonra kolayca tedavi edilir.
Bu hastalıkta kadınlar taşıyıcıdır. Taşıyıcı bir annenin erkek çocuğuna %50 oranında hemofili hastalığını geçirme olasılığı vardır. Hemofili A (faktör VIII eksikliği), yaklaşık her 5.000 erkek doğumunda bir ortaya çıkar ve hemofili olgularının yaklaşık %80 – %85’ini oluşturur. Hemofili B (faktör IX eksikliği) daha nadir olup her 30.000 erkek doğumunda bir görülür.
Teşhis Yöntemi ve Tedavi
Teşhis AH klinik tablodan şüphelenmeli ve anormal bir pıhtılaşma testi ile doğrulanmalıdır. Son zamanlarda anormal kanaması olan ve aktive parsiyel tromboplastin zamanı (aPTT), özellikle yaşlı ve peri ve post-postum kadınlarda izole uzun süreli olan hastalarda tanı düşünülmelidir.
Klinik Test ve Çalışma Rutin birinci satır pıhtılaşma testleri arasında aktive parsiyel tromboplastin zamanı (aPTT) ve protrombin zamanı (PT) bulunur. İki test, iki farklı doku faktörü (aPTT parsiyel tromboplastinde) ile tetiklenen plazmanın koagülasyon süresini ölçer. aPTT, temel olarak FVIII, FIX, FXI ve XII’ye duyarlıdır; PT, karaciğer tarafından sentezlenen pıhtılaşma proteinlerine duyarlıdır (FII, FVII, FIX, “protrombin kompleksi” olarak adlandırılır; K vitaminine bağlı sentez ile).
AH’li bireylerde, normal PT ile izole edilmiş bir uzun süreli aPTT vardır. Spesifik olmayan inhibitörler (örneğin, lupus antikoagülan) veya heparin tedavisi gibi izole uzun süreli aPTT’nin diğer nedenlerini ekarte etmek için testler de yapılır.
Hastanın plazmasını normal plazma ile karıştırmak suretiyle gerçekleştirilen aPTT karıştırma testleri, teşhisi daha da doğrulayabilir. Bir karıştırma çalışması, genetik faktör eksikliklerini faktör inhibitörlerinden ayırmaktadır. Bir kan numunesi alınır ve kontrol grubundan alınan kanla karıştırılır. Faktör eksikliği olan bireylerde normal plazma test değerini normale döndürür; Faktör inhibitörü olan bireylerde yok.
Bir faktör inhibitörü kurulduktan sonra, koagülasyon faktörlerinin (çoğu durumda FVIII) aktivitesini ve inhibitörün titresini ölçmek için bir analiz yapılacaktır. AHA’lı bireylerde bu faktör VIII eksikliği gösterecek ve ciddiyeti de belirleyecektir.
Tedavi AH nadir görülen bir hastalık olduğundan, etkilenen bireyleri tedavi etmek için kullanılan çoğu tedavi raporlarına veya küçük vakalara dayanmaktadır. Spesifik tedavilerin birbirleriyle etkinliklerini karşılaştıran az sayıda çalışma vardır. Sonuç olarak, tedavi oldukça kişiselleştirilmiştir. Spesifik terapötik prosedürler ve müdahaleler, mevcut spesifik semptomları içeren sayısız faktöre bağlı olarak değişecektir; altta yatan neden de dahil olmak üzere hastalığın doğal seyri (biliniyorsa); yaş ve genel sağlık (örneğin eşlik eden hastalık), bazı ilaçların veya prosedürlerin toleransı ve kişisel tercih; ve diğer faktörler. Özel terapötik müdahalelerin kullanımına ilişkin kararlar, doktorun ve sağlık ekibinin diğer üyeleri tarafından, vakasının özelliklerine dayanarak, hasta ve / veya ebeveynleri ile dikkatli istişare içinde verilmelidir; olası yan etkiler ve uzun vadeli etkiler dahil olmak üzere potansiyel fayda ve risklerin kapsamlı bir tartışması; hasta tercihi; ve diğer uygun faktörler.
Spontan remisyon bildirildi; genel olarak doğum sonrası durumlarda (doğumdan sonraki birkaç ay içinde) ve alerjik ilaç reaksiyonuna ikincil durumlarda, genellikle rahatsız edici ilacı bıraktıktan birkaç ay sonra ortaya çıkabilir. Spontan remisyon, düşük titre inhibitörleri olanlar gibi etkilenen diğer bireylerde de görülebilir.
AH’nin yönetimi aşağıdaki hedeflere odaklanır: kanamanın kontrol edilmesi ve önlenmesi (varsa veya önemliyse), inhibitörün ortadan kaldırılması ve altta yatan hastalığın tedavisi (varsa).
Kanama Bölümlerini Kontrol Etme Kanama çok şiddetli olabilir ve ani bir başlangıç olabilir. Bu nedenle morbidite ve mortaliteyi azaltmak için hızlı hemostatik kontrol zorunludur. Uluslararası Tavsiyeler, anti-hemorajik tedavinin, inhibitör titresi ve faktör VIII aktivitesinden bağımsız olarak AH tanısının doğrulandığı ciddi kanaması olan hastalarda başlatılması gerektiğini belirtir. İki yaklaşım vardır: bypass edici ajanların kullanımı (kazanılmış eksikliği atlayan faktörlerin konsantreleri) veya FVIII seviyelerini arttırma stratejilerinin kullanılması. Bu iki seçenek arasındaki seçim, bölgeye ve kanamanın ciddiyetine ve her bir hastanın özelliklerine dayanır. Hemostatik ajanlar öngörülebilir bir etkiye sahip olmadığından, kanama tedavisi bu alanda uzman bir kişi tarafından denetlenmeli ve hemostatik kontrol ve kanama çözünürlüğünü doğrulamak için uygun laboratuvar testleri, görüntüleme teknikleri ve yetenekli bir klinik değerlendirme gereklidir. Fibrin yapıştırıcı veya antifibrinolitik ajanlar, yerel kanamanın kontrolünde faydalı olabilir. Bypassing ajanları, hızlı hareketleri ve yüksek etkinlik seviyeleri nedeniyle önerilen birinci basamak tedavidir. Dozaj büyük ölçüde konjenital hemofili hastalarında FVIII inhibitörleri olan hastaların yönetimi konusundaki deneyime dayanır ve genellikle klinik değerlendirmeye dayanır.
Halen mevcut olan bypass yapan ajanlar, rekombinant aktif faktör VII (rFVIIa veya NovoSeven® RT) veya aktifleştirilmiş protrombin kompleks konsantresidir (aPCC veya FEIBA®). Bu tedavilerin hiçbiri tüm bireylerde etkili değildir.
NovoSeven® RT, faktör VII’nin genetik olarak tasarlanmış (rekombinant) bir versiyonudur. Yapay olarak bir laboratuvarda üretildiği için, insan kanı veya plazması içermez ve sonuç olarak, kan kaynaklı virüsler veya bu gibi diğer patojenlerin riski yoktur. NovoSeven iyi tolere edildi ve çok az yan etkiyle ilişkilendirildi. AH’li bireylerde trombotik yan etki riski (tromboz)% 1’in altındadır. NovoSeven, edinilmiş hemofili hastalarının tedavisinde baypas edici ajan olarak kullanılmak üzere Gıda ve İlaç İdaresi (FDA) tarafından onaylanmıştır.
aPCC, çeşitli aktif pıhtılaşma faktörlerini içeren plazma kaynaklı, anti-inhibitör kompleksidir. Bu faktörler, ilacın kan pıhtılarının oluşumundaki bazı faktörleri atlamasına izin verir (faktör VIII gerektiren basamaklar dahil). aPCC, olası virüsleri veya benzer patojenleri etkisiz hale getirmek için tedavi edilir ve ters trombotik olaylar nadirdir. Şu anda Amerika Birleşik Devletleri’nde mevcut olan aPCC’nin tek şekli, şu anda Shire’ın bir parçası olan Baxalta’dan temin edilebilen FEIBA®’dır. Daha fazla bilgi için iletişime geçin: Baxalta http://www.feiba.com/
FVIII’in, endotel hücreleri tarafından FVIII salınımını indükleyen, FVIII konsantresi veya DDAVP infüzyonu gibi bir FVIII artışına izin veren terapötik yöntemler, inhibitör titresi çok düşük olmadığı sürece (yani <5 Bethesda üniteleri [BU)) ve genellikle yetersiz olarak kabul edilir. atlayan ajanlar mevcut değildir. 2014 yılında ABD FDA, edinilmiş hemofili A (edinilmiş Faktör VIII [FVIII] eksikliği olan yetişkinlerde kanama ataklarının tedavisi için Obizur’ü onayladı. Obizur, şimdi Shire’ın bir parçası olan Baxalta tarafından üretildi. Daha fazla bilgi için: Baxalta http://www.obizur.com/
İnhibitörEradikasyonu
Bazı durumlarda inhibitör, inhibitör bulunduğu sürece kendiliğinden kaybolabilse de, kanamayla ilişkili morbidite ve mortalite önemlidir. Bu nedenle, yetişkinlerde inhibitörün yok edilmesi (iimunosupresyon tedavisi) tedavisinin, açıkça kontrendike olmadıkça AH tanısından hemen sonra başlaması tavsiye edilir. Öneriler, büyük ölçüde, gerçek hayat verilerini toplayan kayıt defterlerinde yapılan gözlemlerden elde edilir.
Genel olarak, tek başına veya siklofosfamid ile ilişkili kortikosteroidler birinci basamak tedavidir. Bu iki yöntemde uzun süreli sağkalımda belirgin bir fark gözlenmedi. Bununla birlikte, bireyler immünosüpresif ilaçlara farklı tepkiler verir ve bir bireyde etkili olan diğerinde etkili olmayabilir. Siklosporin A, azatiyoprin, vinkristin, mikofenolat mofetil ve 2-klorodeoksiadenozin de dahil olmak üzere edinilmiş hemofili tedavisi için çeşitli ek immünosüpresif maddeler kullanılmıştır.
Tedaviye cevap kriterleri henüz belirlenmemiştir; Bununla birlikte, yüksek inhibitör titresi ve düşük FVIII düzeyi tedaviye yanıtı öngörüyor gibi görünmektedir.
AH’nin nüksetmesi, immünosüpresif tedavi durdurulduktan sonra veya doz azaltıldığında remisyon sağlayan kişilerde ortaya çıkabilir. İlişkili yan etkiler nedeniyle, uzun süreli immünsüpresif tedavi önerilmemektedir.
AH’li bireylerin, inhibitör eradikasyonundan sonraya kadar önemli bir travma riski taşıyan etkinliklerden kaçınmaları teşvik edilir.
AH’li hastalar federal olarak finanse edilen hemofili tedavi merkezine yönlendirmeden faydalanacaktır. Bu ihtisas merkezleri hemofili hastalığına ve spesifik tedavi planlarının geliştirilmesi, etkilenen bireylerin izlenmesi ve takibi ve son teknoloji tıbbi bakım da dahil olmak üzere ilgili hastalıklar için kapsamlı bakım sağlayabilir. Bir hemofili tedavi merkezinde tedavi, bireylerin ve aile üyelerinin, hemofili hastalarının tedavisinde deneyimli profesyonel bir sağlık ekibi (doktorlar, hemşireler, fizyoterapistler, sosyal hizmet uzmanları ve genetik danışmanlar) tarafından korunmalarını sağlar.
Araştırma Terapileri Araştırmacılar, AH’li bireyler için potansiyel bir terapi olarak, rituximab ilacını inceliyorlar. Bu ilaç, hastalığa neden olan otoantikorlara saldırır. Rituximab, monoklonal bir antikor veya biyolojik terapi olarak sınıflandırılır – antikorlar gibi hareket eden ilaçlar, ancak yapay olarak bir laboratuvarda yaratılırlar. Başlangıçta, rituximab inhibitörlerin yok edilmesinde ümit verici sonuçlar göstermiştir. Bazı araştırmacılar, kortikosteroid / siklofosfamid tedavisine cevap vermeyen bireylerde inhibitörlerin yok edilmesi için ikinci basamak bir tedavi olarak veya kortikosteroid / siklofosfamid tedavisini tolere edemeyen bireylerde birinci basamak tedavi olarak rituximab önermişlerdir. AH tedavisi için rituksimabın uzun vadeli emniyetini ve etkinliğini belirlemek için daha fazla araştırma gereklidir.
AH’de inhibitörleri yok etmek için yüksek dozda intravenöz immünoglobulin kullanılmıştır. Bununla birlikte, tıbbi literatürdeki çoğu rapor hayal kırıklığı yaratan sonuçları detaylandırmaktadır. Bazı araştırmacılar, bu tedavinin, inhibitörleri yok eden diğer ilaçlara veya prosedürlere yardımcı bir tedavi olarak en iyi şekilde söz verdiğine inanmaktadır.
Yüksek inhibitör titresi ve şiddetli kanamaya sahip bazı kişiler, plazmaferez veya immüno-emilim adı verilen bir prosedüre maruz kalabilir. Bu prosedürler genellikle diğer tedavi seçeneklerine cevap vermeyen ve hayatı tehdit edici kanama bölümleri yaşayan hastalar için ayrılmıştır. Plazmaferez, istenmeyen maddelerin kandan uzaklaştırılmasını içerir. Kan hastadan çıkarılır ve katı kan hücreleri sıvı plazmadan ayrılır. Hastanın plazması daha sonra, orijinal kan hücreleri ile birlikte yeniden transfekte edilen donör insan plazması veya albümini ile değiştirilir. Modifiye Bonn-Malmö Protokolü (MBMP), immünoglobulinleri (FVIII inhibitörü) FVIII replasmanı ve immünosüpresyonu ile spesifik olarak bağlayan kolonlarda immü emilimini birleştirir ve akut kanamanın hızlı ve güvenli kontrolünü sağlayabilir. Esas olarak Avrupa’da uygulanmaktadır.
İmmünsüpresif tedavinin komplikasyonları (IST) IST zaten yaşlı, kırılgan popülasyonda enfeksiyon riskini arttırır. Nötropeni (beyaz kan hücrelerinin azalması) ve sepsis sıklıkla bildirilir ve ölüme katkıda bulunur. Yükseltilmiş kan şekeri düzeyleri, gastrointestinal ülserler, kas bozuklukları ve psikiyatrik bozukluklar (% 3) dahil olmak üzere diğer iyi bilinen steroid tedavisi komplikasyonları bildirilmiştir.
Sonuçlar, Araştırma ve Diğer İhtiyaçlar AH, hastayı değerlendiren ilk kişi olan internist veya acil servis doktoru tarafından daha sık karşılaşılır. AH’nin gerçek dünyadaki klinik uygulamalarda tanı konmaması ve yanlış tanı konması muhtemeldir, bu durumun sağlık hizmetleri pratisyenleri arasında bu hastalığa ilişkin farkındalık yaratması ve AH’nin yönetimindeki uzmanlara yönlendirmeyi teşvik etmesi gerektiğini gösterir. Birçoğu için, AH, derhal tanınması ve uygun yönetimin net faydaları ile büyük ölçüde tedavi edilebilir. Hemostatik hem de immün tedavi, önemli riskler getirir ve güvenliği sağlamak için yakın izleme gerektirir. AH araştırmalarında en yüksek öncelik, tedavinin yan etkileri ile daha iyi dengeleyici inhibitör eradikasyonu ile sağkalımı artıran daha güvenli immünsüpresif rejimler geliştirmektir.
Hastalıkla İlişkili Genler İlgili Bozukluklar
Aşağıdaki bozuklukların belirtileri AH’ninkine benzer olabilir. Karşılaştırmalar ayırıcı tanı için faydalı olabilir. Hemofili, bir grup nadir kanama bozukluğu grubu için genel bir terimdir. Hemofili hastalarının çoğu, inaktif veya eksik kan proteinlerinin neden olduğu kan pıhtılaşması (pıhtılaşma) bozukluğudur. Üç ana kalıtsal hemofili şekli vardır: hemofili A (aynı zamanda klasik hemofili, faktör VIII eksikliği veya antihemofilik globulin [AHG] eksikliği olarak da bilinir); hemofili B (Noel hastalığı veya faktör IX eksikliği); ve hemofili C (faktör XI eksikliği). Hemofili A ve B, X’e bağlı resesif genetik bozukluklar olarak kalırken, hemofili C, otozomal resesif genetik hastalık olarak kalıtılır. Hemofili A ve B çoğunlukla erkeklerde ifade edilir, ancak kadınlar da etkilenebilir. Hemofili C erkekleri ve dişileri eşit sayıda etkiler. Hemofili hafif, orta veya şiddetli olarak sınıflandırılabilir; şiddeti seviyesi kandaki aktif pıhtılaşma faktörü yüzdesiyle belirlenir (normal yüzde 50 ila 150 arasında değişmektedir). Şiddetli hemofili hastaları kanlarında aktif pıhtılaşma faktörünün yüzde birinden daha azına sahiptir. (Bu hastalıklar hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “hemofili” yi seçin.)
Von Willebrand hastalığı (VWD), uzun süreli kanama ile sonuçlanan kalıtsal bir kanama bozukluğudur ve etkileri bakımından geniş ölçüde değişkenlik gösterir. VWD’li bireylerde von Willebrand faktörü eksikliği veya eksikliği vardır. Ayrıca, VIII faktörü olarak bilinen düşük bir ilave faktör seviyelerine sahip olabilirler. Willebrand faktörü eksik veya kusurlu, trombositlerin yanlış işleyişi, hemostazın erken dönemlerinde yer alan özel kan hücresi fragmanları, kanamayı durdurmak için damar duvarı lezyonu bölgesinde tıkaç oluşumu ile sonuçlanır. VWD’li bireylerde trombositler kan damarlarındaki deliklere yapışmaz ve kanama uzar. Çoğu insan nispeten hafif bir hastalığa sahiptir ve yetişkin olana kadar teşhis edilmez. Bireylerin küçük bir yüzdesi, bebeklik döneminde veya erken çocukluk döneminde, uzun süreli kanama ve anormal derecede yavaş bir pıhtılaşma süresi gibi problemler yaşamaya başlar. Belirtileri gastrointestinal kanama, burun kanamaları, diş eti kanaması ve kolay morarma olabilir. Etkilenen kişiler, yaralanma, doğum ve / veya ameliyattan sonra kolayca kanabilir. Hastalığın üç ana formu vardır. Otozomal dominant bozukluk olarak iki tip kalıtımsaldır; üçüncü tip otozomal resesif bir hastalık olarak kalıtsaldır. (Bu hastalık hakkında daha fazla bilgi için, Nadir Hastalık Veritabanında arama teriminiz olarak “von Willebrand” ı seçin.)
Hastalığın Diğer İsimleri ⁃ Kazanılmış (Edinilmiş) Hemofili ⁃ Edinsel Hemofili ⁃ AH ⁃ AHA – AHB
11q24.2
lokasyonundaki HYLS1 geninin
mutasyonuna bağlı olarak ortaya çıkan ve otozomal resesif aktarılan bu sendrom
ilk olarak Finlandiya’da yaygın olan Meckel Sendromu üzerine çalışılırken
keşfedilmiştir. Hidrolatalus da tıpkı Meckel sendromu gibi polidaktili[1] ve santral sinir sistemi
malformasyonu ile karakterizedir. Ancak Meckel sendromundaki polikistik böbrek
ve karaciğer bu sendromda görülmez. Santral sinir sisteminde ise Meckel
sendromundaki gibi ensefalosel[2] görülmez bunun yerine
hastalarda hidrosefali[3] vardır. Embriyonik dönemde
sıklıkla masif polihidroamniyos[4] vardır ve bu da preterm
eyleme [5]neden olur. Ventriküller
subaraknoid boşluğa açıldığı için eksternal hidrosefalus görülür. Foramen
magnum anahtar deliği görünümündedir. Polidaktili elde postaksiyel(ulna
tarafında) ayakta preaksiyeldir(tibia tarafında). Halluks dubleks [6]ve pes ekinovarus(clubfoot)[7] yaygın görülen
deformitelerdir. Mandibula [8] küçük, gözler ve burun az
gelişmiştir. Hastaların yarısında büyük atriyoventriküler comünis defekti [9] vardır. Buna anormal
akciğer lobları ve havayolu darlığı da eşlik eder. USG ile prenatal tanı mümkündür. (McKusick,
1986)
(ScienceDirect, 1999)
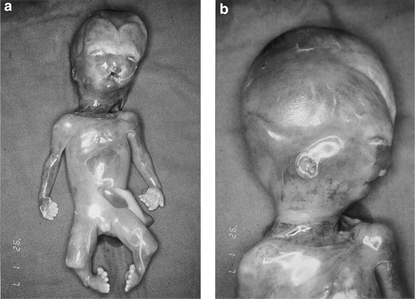
Şekil 1 Termine edilmiş 18 haftalık gebelik ürünü hidrolatalus sendromlu erkek fetüs. Hidrosefali, küçük ve uzak yerleşmiş gözler, bifid (çift tepeli) burun, çift taraflı yarık dudak, mikrognati (küçük alt çene), düşük kulaklar, polidaktili, kısa bacaklar ve yumru ayak.
McKusick, V. A.
(1986, Haziran 3). Online Mendelian Inheritance in Man:OMIM Web Sitesi:
https://www.omim.org/entry/236680#clinicalFeatures adresinden alınmıştır
[1] Doğuştan el ya da ayaklarda
normalde olması gerekenden daha fazla parmak bulunması
[2] Beyin dokusunun kafatasındaki bir
açıklıktan dışarıya çıkması
[3] Beyinde sıvı toplanması sonucu
kafanın anormal büyümesi
[4] Rahim içerisinde bebeğin içinde
bulunduğu amniyon sıvısının normalden fazla olması
Anensefali, beyin ve kafatası kemiklerinin anormal
gelişimi ile karakterize bir nöral tüp defektidir. Nöral tüp, normal bir
gebeliğin 3. Ve 4. Haftaları arasında kapanan ve bununla beraber embriyonun
beynini ve omuriliğini oluşturan dar bir geçittir. Nöral tüpün kafayı oluşturan
kısmı kapanamadığında, beynin, kafatasının ve üstünü örten saçlı derinin büyük
bir kısmının yokluğu Anensefali olarak adlandırılır. Bu hastalığa sahip
bebekler, önbeyin ve serebrum olmadan doğarlar. Bu eksiklikler genellikle bu
bebeklerin görme ve işitme engelli olmalarına, bilinçten ve acı hissetme
yetisinden yoksun olmalarına sebep olur. Anensefaliye sahip neredeyse her bebek
anne karnında ölmektedir. Bazı bebekler doğumdan sonra birkaç saat veyahut
birkaç gün yaşayabilmektedir.
Genetik
Değişikler/ Etken Faktörler
Anensefalinin altında yatan nedenler henüz tam olarak
anlaşılamamıştır. Diğer nöral tüp defektleri gibi anensefalinin de birden fazla
genle ve çevresel faktörle alakalı olduğu düşünülmektedir.
MTFHR geni, vücutta Folat (Vitamin B9) üretimini
sağlayan gendir. Bu vitaminin eksikliği nöral tüp defektlerinden sorumlu bir
etkendir. Gebelikten önce ve gebelik süresince folik asit takviyesi almak nöral
tüp defektlerini azaltmakta yardımcı olmaktadır.
Aynı zamanda gebede diyabet varlığı, obezite varlığı,
yüksek sıcaklıklara uzun süre maruz kalma (ateşli hastalık/sauna/hamam), belli
başlı nöbet önleyici ilaçlar nöral tüp defekti oluşma riski meydana getirirler.
Belirti
ve Semptomlar
Anensefalik
kafatası görünümü
Anensefaliye
sahip neonatlarda ayrıca renal yetmezlik ve spina bfida da sık izlenir.
Genetik
Görülme Sıklığı
Anensefali oldukça sık görülen bir nöral tüp
defektidir. Sıklığı 1000 gebelikte 1’dir.
Kalıtım
Paterni
Anensefalide sporadik vakalar izlenir. Bunun anlamı
hastalığa sahip olan hastalarda aile öyküsüne rastlanılmamasıdır. Kalıtım
paterni belirsizdir.
Teşhis
Yöntemleri/ Tedaviler
Prenatal tanı mümkündür, tedavi bulunmamaktadır.
Hastalığın prognozu oldukça kötü olduğundan gebelikler genellikle abortus
(düşük) ile sonuçlanır.
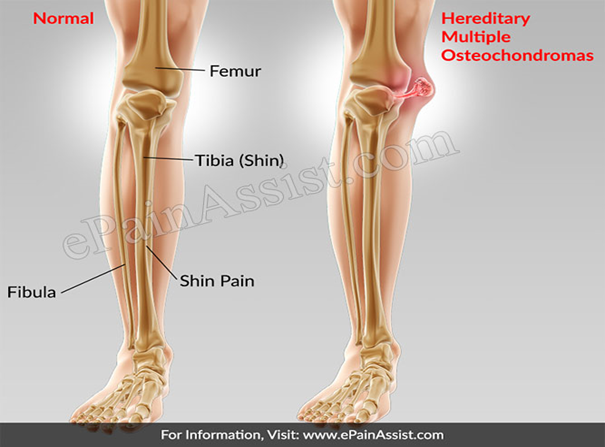
Genel Bilgi Kalıtsal multipl osteochondromas (HMO) , insanların osteochondromas adı verilen multipl benign (kanserli olmayan) kemik tümörleri geliştirdiği bir durumdur. Osteokondromların ve bulundukları kemiklerin sayısı etkilenen bireyler arasında büyük ölçüde değişiklik gösterir. Osteokondrom doğumda mevcut değildir, ancak etkilenen kişilerin yaklaşık yüzde 96’sı 12 yaşına geldiğinde çoklu osteokondrom geliştirir. Osteokondromlar tipik olarak uzun kemiklerin sonunda oluşur. ve kalça ve omuz bıçağı gibi yassı kemiklerde. (Görsel 1)
Çoklu osteokondromlar
kemik büyümesini bozabilir ve kolların, ellerin ve bacakların büyümesine neden
olarak kısa boylanmaya neden olabilir. Genellikle kemik büyümesiyle ilgili
bu problemler sağ ve sol ekstremiteyi eşit şekilde etkilemez, eşit olmayan
ekstremite uzunlukları (ekstremite uzunluğu tutarsızlığı) ile sonuçlanır. Önkol
veya ayak bileğinin eğilmesi ve osteokondromların neden olduğu kalça
eklemlerinin (kalça displazisi) anormal gelişimi yürüme zorluğuna ve genel
rahatsızlığa neden olabilir. Çoklu osteokondromlar ayrıca ağrı, sınırlı
eklem hareket aralığı ve sinirler, kan damarları, omurilik ve osteokondromları
çevreleyen dokular üzerinde baskı ile de belirti gösterebilir.
Osteokondromlar tipik
olarak benigndir; Bununla birlikte, bazı durumlarda bu tümörler malign
hale gelir (kanserli). Araştırmacılar, kalıtsal multipl
osteokondromu olan kişilerin 200 yaşamda 1 ila 20’de 1 oranında
kanserli osteokondrom (sarkomlar) gelişme riski olduğunu tahmin
etmektedir .
Kalıtsal çoklu
osteokondromlar eskiden kalıtsal çoklu exostoses olarak biliniyordu. Exostoses
küçükse, hasta üzerinde etkisi çok az olabilir veya çok az olabilir, ancak daha
ciddi vakalarda, büyüme önkol, diz, ayak bilekleri, omurga ve / veya pelvis
deformitelerine neden olabilir. Buna karşılık, bu deformasyonlar, harekete
veya dolaşıma müdahale eden ve ciddi ağrıya neden olabilen sinirlere,
tendonlara ve kan damarlarına dayayabilir. Yürüme zorluğu ve genel
rahatsızlık da yaygındır. Bu komplikasyonlara ek olarak, etkilenen bazı
bireylerin kısaltılmış ve / veya eğilmiş bacaklarından hafif kısa boyları,
uyuşukluk ve / veya felçten kaynaklanan omurilik sıkışması veya pelvik bölgede
ise idrar tıkanması vardır.
Genetik Değişiklikler / Etken Faktörler
HMO, EXT1 ve EXT2
genlerindeki HARICI1 ve HARICI2 mutasyonların neden olduğu
kalıtsal osteokondromlardır. HARICI 1 geni ve HARICI2 gen,
sırasıyla, exostosin-1 ve exostosin-2 proteinlerinin üretilmesine yönelik
talimatlar sağlar. İki ekzostosin proteini birbirine bağlanır ve Golgi aygıtı olarak adlandırılan bir
hücre yapısında bulunan bir kompleks oluşturur, yeni üretilen enzimleri ve
diğer proteinleri değiştirir. Golgi aparatında, exostosin-1 ve exostosin-2
kompleksi, heparan sülfat adı verilen bir proteini modifiye eder, böylece hücre
tarafından kullanılabilir.
Exostosin-1 veya
exostosin-2’de bir mutasyon olduğunda, heparan sülfat doğru işlenemez ve
işlevsel değildir. Heparan sülfat birçok vücut işleminde yer almasına
rağmen, bu proteinin eksikliğinin osteokondrom gelişimine nasıl katkıda
bulunduğu açık değildir. Araştırmacılar, kalıtsal çoklu
osteokondromlu kişilerin yaklaşık yüzde 15’inin EXT1 veya EXT2geninde mutasyon olmadığını tahmin etmektedir. Bu bireylerde
neden çoklu osteokondromların oluştuğu bilinmemektedir.
Belirti ve Semptomlar
Kalıtsal multipl
exostoz olarak da adlandırılan kalıtsal multipl
osteokondromlar (HMO), çoklu kıkırdak kaplı gelişimine neden olan genetik
bir hastalıktır. Tümörler, kemiklerin dış yüzeylerinde (osteokondromlar) gelişim gösterir. Osteokondromlar
tipik olarak çocukluk veya ergenlik döneminde belirgin hale gelir ve
osteokondromların sayısı, büyüklüğü ve yeri kişiden kişiye
değişir. Belirti ve semptomlar arasında ağrı, azalmış hareket açıklığı,
sinir sıkışması, deformite, uzuv uzunluklarındaki farklılıklar, kısa boy ve
kırıklar sayılabilir. Kaburgaların osteokondromları çökmüş bir akciğer (pnömotoraks), hemotoraks veya perikardiyal efüzyon gibi
komplikasyonlara neden olabilir. Osteokondromlar tipik olarak çocukluk
boyunca büyür ve büyüme plakaları kapandığında büyümeyi durdurur. Bununla
birlikte, bazı insanlarda daha sonra tekrar edebilirler. Osteokondromların
büyük çoğunluğu iyi huyluyken (kanserli olmayan), erişkinlikte HMO’lu
kişilerin% 2 ila% 5’inde malign hale gelebilir (kanserli).
Genetik Görülme Sıklığı
Kalıtsal multipl
osteokondromların görülme sıklığının 50.000 kişide 1 olduğu tahmin
edilmektedir. Bu durum bazı izole edilmiş popülasyonlarda daha sık
görülür: insidans Guam’ın Chamorro popülasyonunda yaklaşık 1.000’de 1 ve Kanada’nın
Manitoba kentindeki Ojibway Hint popülasyonunda 77’de 1’dir.
Kalıtım Paterni/Deseni
Bu durum otozomal dominant paternde kalıtsaldır; bu,her
hücrede değiştirilmiş genin bir kopyasının, hastalığa neden olmak için yeterli
olduğu anlamına gelir.
Teşhis Yöntemleri ve Tedavileri
Teşhis
Etkilenen bölgenin x-ışını , MRI veya BT taramaları şeklinde yapılan radyografik çalışmalarla birlikte yapılan kapsamlı bir klinik değerlendirme , bunun teşhisine işaret edebilir. Ek olarak, bir moleküler test, tanısını doğrulayacak olan EXT1 veya EXT2 genindeki mutasyonları açıkça gösterecektir.
Tedavi
Osteokondromların yerlerine ve boyutlarına ve neden oldukları semptomların şiddetine bağlıdır. Tedavi seçeneklerine örnek olarak “izle ve bekle” yaklaşımı (semptom olmadığında), tümörün cerrahi olarak çıkarılması, düzeltici osteotomi ve büyüme plakası durması veya uzuv uzatma prosedürleri verilebilir. İyi huylu osteokondromlar genellikle yaşam beklentisini etkilemese de, yine de çeşitli sağlık sorunlarına ve yaşam kalitesini bozabilecek semptomlara neden olabilir. Malign hale gelen osteokondromlar (kondrosarkomlara veya osteosarkomlara dönüşmek) hayatı tehdit edici olabilir, ancak bu vakalardaki görünüm tümör derecesine bağlı olabilir .
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}