Hiper-Ig E Sendromu bağışıklık sisteminde
yetmezliğin yol açtığı semptomlarla seyreden nadir görülen bir hastalıktır.
Anne-babadan otozomal baskın ya da otozomal çekinik olarak aktarılabilir veya hastalıklı bireyde yeni
bir mutasyonun oluşmasıyla ortaya çıkabilir.
Bu hastalığın otozomal baskın kalıtımıyla
ortaya çıkan formu olan Otozomal Baskın Hiper-IgE Sendromu (OB-HIES) diğer bir
adıyla Job Sendromu, vücutta başlıca bağışıklık sistemini etkilemektedir. Bu
hastalarda tekrarlayan enfeksiyonlar (zatürre, cilt enfeksiyonları), egzama,
kemik ve diş anormallikleri görülür. Hastaların kanında İmmunglobulin E (IgE)
antikoru yüksektir. (1)
Genetik
Değişiklikler/Etken Faktörler
Vakaların çoğunda STAT3 genindeki mutasyon
hastalığa sebep olmaktadır. Bu genin kodladığı STAT3 proteini, başta T
hücreleri olmak üzere bağışıklık sistemindeki hücrelerin olgunlaşmasında rol
oynayan bir proteindir. Bağışıklık sistemi hücreleri normalde bakteri ve mantar
gibi hastalık yapıcı etkenlere karşı vücut savunmasını sağlar.
STAT3 genindeki mutasyonlar STAT3 proteininin
yapı ve fonksiyonlarında değişikliklere yol açar. Sonuç olarak bağışıklık
sisteminde anormallikler oluşur ve bu da vücudu, özellikle de akciğer ve cilt
dokusunu, bakteri ve mantar enfeksiyonlarına duyarlı hale getirir. STAT3
proteini ayrıca kemik dokusundaki yapım ve yıkım olaylarına da katılır, bu
yüzden kemik ve dişlerde anormallikler görülmektedir. STAT3 gen mutasyonunun
IgE seviyelerini hangi mekanizmayla yükselttiği bilinmemektedir. (2)
Belirti
ve Semptomlar
OB-HIES yenidoğan döneminde döküntülerle
ortaya çıkar ve bağışıklık sistemini, kemikleri, bağ dokusu ve diş gelişimini
etkiler. Ciltte egzama ve tekrarlayan enfeksiyonlar görülür, bu enfeksiyonlar
içi iltihap dolu apseler oluşturur. Tekrarlayan zatürre gibi akciğer
enfeksiyonları akciğerde içi hava dolu keselerin oluşmasına neden olur
(Pnömatosel). Tekrarlayan solunum yolu enfeksiyonları kronik solunum
yetmezliğine yol açabilir. Bu hastalarda Candida (mantar) enfeksiyonları da sık
görülmektedir. Hastalarda tipik bir yüz görünümü vardır: Pürüzlü cilt, yüzde
asimetri, alın ve çenede çıkıklık, gözlerde çukurluk, geniş burun köprüsü ve
dolgun burun ucu. Hastalarda tekrarlayan kemik kırıkları ve skolyoz
görülebilir; diş yapımında bozukluklar vardır. Anevrizma, tromboz ve PDA gibi
damarsal bulgular da bildirilmiştir. Gözde ksantelezma, arpacık, göz kapağı
nodülleri, şaşılık ve retina dekolmanı gibi komplikasyonlar olabilir.
Hastalarda otoimmün ve lenfoproliferatif hastalık görülme riski artmıştır. (3)
Görsel 1: OB-HIES tipik yüz görüntüsü (4)
Genetik
Görülme Sıklığı
OD-HIES nadir bir hastalıktır ve görülme
sıklığı 1/1.000.000’dur. (3)
Kalıtım
Paterni
Bu hastalık otozomal baskın kalıtılır, yani
mutasyonlu geni taşıyan herkes hastalığa sahiptir. Hastaların yaklaşık yarısında
mutasyonlu gen, etkilenmiş anne ya da babadan kalıtılır yani tek bir ebeveynin
hastalıklı geni çocuğa aktarması, hastalığın oluşumu için yeterlidir. Diğer
vakalarda ise anne ya da babada hastalık yoktur, hastalığın sorumlusu gende
oluşan yeni mutasyonlardır. (2)
Teşhis
Yöntemleri ve Tedaviler
Tanıda klinik bulgularla beraber laboratuvar
bulguları değerlendirilir. Kanda 2000 U/ml’nin üstündeki IgE değerleri
hastalığın tanısı için anlamlıdır, hatta çoğu zaman IgE değerleri 5000 U/ml’den
de yüksek görülür. STAT3 mutasyonunun genetik olarak gösterilmesi tanıyı
doğrular.
Bu hastalığın kesin bir tedavisi yoktur,
tedavi yaklaşımı enfeksiyonların önlenmesine yöneliktir, bu amaçla uzun dönemli
antibiyotik ve antifungaller (mantar ilacı) kullanılır. OB-HIES yaşam boyu
süren kronik bir hastalıktır ve oluşan her yeni enfeksiyon tedavi edilmelidir.
Akciğer apseleri için cerrahi müdahale gerekebilir. (3) Egzama için
nemlendiriciler ve kullanımı sınırlı tutulmakla birlikte topikal steroidler
kullanılabilir. Apselerin tedavisi drenajdır (apsenin boşaltılması). Kemik
iliği nakli Otozomal Resesif Hiper IgE Sendromu için tedavi edici olsa da,
OB-HIES tedavisinde önerilmemektedir. (5)
Hastalıkla
İlişkili Genler
17. kromozomun uzun kolunda lokalize olan
STAT3 geni vakaların büyük bir kısmından sorumludur. STAT3 gen mutasyonu
olmayan kişilerde hastalığın genetik nedeni bilinmemektedir. (2)
Larsen sendromu LRS vücutta kemiklerde anormal
büyümeye yol açan otozomal dominant nadir bir genetik hastalıktır. Sendrom ilk
olarak 1950 yılında yayınlanan bir dergide (MD), Amerikan ortopedist Loren J. Larsen tarafından
tarif edilmiştir ve ismini “Larsen” olarak almıştır. Her yıl yaklaşık
100.000 bebekten 1’ini etkileyen sendrom, aynı ailedeki çocuklarda bile,
bozukluğu olan birçok farklı semptomlara neden olabilir. LarsenSendromlu kişiler normal bir zekaya sahiptir. (1) Belirtiler esas
olarak eklemlerden ve iskeletlerden gelir. Bozukluğun karakteristik bulguları
arasında büyük eklemlerin çıkıkları, iskelet malformasyonları, kısa boy ve
ayırt edici yüz, bacak özellikleri bulunur. Sendrom kalıtsaldır ve tüm
vücut dokularının gelişimi için önemli olan bir protein eksikliğinden
kaynaklanmaktadır. Bu protein bağ dokusu proteini filamin B(FLNB)’dir ve
sendrom buproteini kodlayan gendeki mutasyonlardan
kaynaklanır. Mutasyon kendiliğinden ortaya çıkabilir veya otozomal
dominant özellik olarak kalıtsal olabilir. (3) Tedavi temel olarak eklem
kusurlarının düzeltilmesi ve önlenmesinden oluşur. Çoğu insan için bu sendrom,
günlük yaşamda veya kariyer seçimlerinde önemli bir sınırlamaya neden olmaz.
(2) Aşağıda Larsen Sendromlu bireylerin ayırt
edici yüz özellikleri görülmektedir.
Şekil 1. Larsen Sendromu tanısı olan hastaların yüz özellikleri. (5)
Görülme Sıklığı
Larsen Sendromu, erkekleri ve kadınları eşit düzeyde
etkiler. Genel popülasyondaki 100.000 kişiden 1’inde meydana geldiği
tahmin edilmektedir. Larsen Sendromu tanısındaki zorluk nedeniyle, genel
popülasyondaki gerçek sıklığını belirlemek zordur. (3) Orphanet
epidemiyolojik verilerine göre ise bu değer Avrupa’da 1-9 / 1000000 şeklinde verilmiştir.
Belirti ve Semptomlar
Larsen Sendromlu
bireylerde normalden daha geniş gözler (hipertelorizm), belirgin bir alın ve
çökük köprülü burun gibi kendine özgü yüz özellikleri bulunur. Ağız çatısının
(yarık damak) ya da boğazın arkasına sarkan yumuşak dokudaki yarığın (bifid
uvula) tam olarak kapanmaması da etkilenen bireylerin %15’inde ortaya
çıkabilir. Sağırlık genellikle kulak çınlaması (kulak çınlaması) ile
yaygındır ve bireylerin %21’inde orta kulak hücrelerinin malformasyonları ile
ilişkili olabilir. Klasik Larsen Sendromu olan az sayıda bireyde, trakeomalazi
(soluk borusunun kıkırdakta anormal yumuşaması) bulunur. (3) Bu semptomlar aynı
aile bireyleri içinde bile büyük ölçüde değişmektedir. Aşağıda Larsen
Sendromu’nun genel belirti ve semptomları sıralanmıştır. (2)
• Skolyoz veya kifoz gibi omurilik deformasyonu ve servikal omurga
anormallikleri
• Çıkık kalçalar, dizler ve dirsekler
• Anormal derecede gevşek eklemler
• Bilekte ve ayak bileklerinde ekstra
kemikler
• Yassı, kare şeklindeki parmak uçları
• Öne çıkan bir alın, burnun yassı köprüsü,
geniş gözler ve yarık damak gibi kraniyofasiyal anomaliler
• İşitme kaybı (kulaklardaki bazı kemikler
düzgün şekilde oluşmamıştır)
Kalıtım Deseni Larsen Sendromu otozomal dominant kalıtılır ve bu hastalığa kromozom 3’ün (3p14.3) kısa kolunun üzerinde bulunan FNLB adlı gendeki mutasyon sebep olur. FLNB’dekimutasyonlar, bu gen tarafından kodlanan proteinin fonksiyon bozukluğuna neden olur.Filamin B( FLNB ), aktin hücre iskeleti filamentlerini çapraz bağlayan büyük bir dimerik aktin bağlama proteinidir. Bu protein, hücrelerin şeklinin ve hacminin korunmasına yardımcı olur. Filamin B, erken fetal gelişim sırasında kas hücre liflerine bağlanarak kas oluşumunda rol oynar (embriyonik kas hücresi). Ayrıca protein filamenti A’ya (FLNA) bağlanarak, ilkel sinir hücrelerinin farklı bölgelere göç etmesini sağlar.(2)
Otozomal dominantta
mutasyona uğramış genden her bir hücrede bir kopya olması belirtilerin
görünmesi için yeterlidir. Bazı vakalarda hastalıktan etkilenen kişi bunu
mutasyondan etkilenen ebeveyninden alır. Bazı durumlarda da mutasyon
kendiliğinden ortaya çıkar.
Bazı araştırmacılar
bu hastalığın otozomal resesif kalıtıldığını, etkilenmemiş ebeveyne ve
etkilenmiş kardeşe sahip bireyin bu hastalıktan germline mozaizm (gonadal
mozaiklik) sayesinde etkilenmediğini öne sürmektedir. Yani çok çocuklu
ailelerde hasta olmayan ama yumurta ya da spermlerinde (diğer vücut
hücrelerinde değil) mutasyon görünen ebeveyn hastalık yapıcı mutasyon
kalıtılabilir. Bu da hastalığın otozomal resesif görünmesine neden olabilir.
(3)
Otozomal resesif formda Larsen Sendromu, Afrika’nın doğu kıyılarındaki Reunion Adası’ndaki birçok büyük ailede tespit edilmiştir. Bu bozukluk da çoklu eklem çıkıklarına neden olur ancak farklı klinik ve radyolojik özelliklere sahiptir. B4GALT7’de bulunan bir mutasyondan kaynaklanmaktadır. Otozomal resesif Larsen Sendromu olan hastalarda karbonhidrat sülfotransferaz 3 (CHST3 ) genindeki mutasyonlar tanımlanmıştır. Homozigoz GZF1 varyantları şiddetli miyopi, retina dekolmanı ve tekrarlayan büyük eklem çıkıklarından etkilenen Suudi ailelerinde de bu durum bildirilmiştir. (4)
Hastalıkla İlişkili Genler
Larsen Sendromu ile
ilişkili genler; FlnB (filamin B), FlnA (filamin A), CHST3 (karbonhidrat
sülfotransferaz 3), GZF1 (çinko parmak proteini 1), COL7A1 (kollajen tip 7 alfa
1 zinciri), B4GALT7 (beta 1-4 galaktosiltransefarz 7) olarak gösterilmiştir.
(4)
Teşhis ve Tedavi Yöntemleri
Larsen Sendromu’nun
tanısı, ayrıntılı bir klinik değerlendirmeye, ayrıntılı hasta geçmişine ve
karakteristik klinik ve radyolojik bulguların tanımlanmasına dayanarak
yapılır. Radyografik muayene ile iskelet malformasyonları tespit edilebilir. Moleküler
genetik testler FLNB gen mutasyonunun varlığını doğrulayabilir. Teşhis ile
bağlantılı olarak, aileye genetik rehberlik yapılması önemlidir. Ayrıca aynı
sendromlu daha fazla çocuğa sahip olma olasılığının değerlendirilmesi de
gerekir. Ailedeki mutasyon biliniyorsa, birçok kalıtsal hastalık için,
preimplantasyonal genetik tanılama (PGD) yapılmalıdır. Aşağıda Larsen Sendromu
teşhisi için kullanılan yöntemler sıralanmıştır. (2)
• Vücuttaki organ ve yapıların ayrıntılı
görüntüleri için büyük mıknatıslar, radyo frekansları ve bir bilgisayar
kombinasyonu kullanan manyetik rezonans görüntüleme (MRG).
• Vücudun enine kesit görüntüleri için X
ışınları ve bilgisayar teknolojisinin bir kombinasyonunu kullanan bilgisayarlı
tomografi (CT) taraması.
• İki düzlemsel görüntüden 3 boyutlu modeller
oluşturan görüntüleme teknolojisi EOS görüntüleme. (CT taramasından farklı
olarak, çocuk dik veya ayakta dururken EOS görüntüleri alınır ve bu da ağırlık
taşıyan konumlandırma nedeniyle gelişmiş tanı sağlar)
• İlaç kullanımı ve etkinliğini, biyokimyasal
hastalıkları ve organ fonksiyonunu belirlemeye yardımcı olabilecek kan
testleri.
Larsen Sendromu
birçok vücut sistemini etkileyen bir hastalıktır. Dolayısıyla birçok tıbbi
bölümün multidisipliner çalışması gereklidir. Omurga, kemik ve kasla ilgili
sorunlar için ortopedist, düzenli işitme değerlendirmeleri için kulak burun
boğaz uzmanı, yarık damak için plastik cerrah, herhangi bir sinir sorunu için
nörolog, solunum sorunları için pulmonolog, çocuğunuzun fiziksel becerisini geliştirmek
için fizyoterapistler gerekebilir. Fizik tedavi gibi cerrahi olmayan
seçenekleri veya büyümeye devam ederken omurgayı sabitlemek için spinal füzyon
veya büyüyen çubuklar yerleştirmek gibi cerrahi seçenekler kullanılabilir.
Bunun yanında kalça ve diz protezi ihtiyaçları olabilir. (2)
Özellikle yeni doğan
bebeklerde, ayakların birkaç ay sonra ameliyat edilmesi gerekir, ancak
başlangıçta tedavi, ayakları kademeli olarak daha iyi bir pozisyona
getirebilmek için tekrarlanan germe ve sıva işlemlerinden oluşur. Daha
sonra, işlem bir ray (ortez) ile gerçekleştirilir. Bu tedaviye rağmen,
ayak hatası ayarının bozulma riski veya optimum pozisyonun elde edilememesi
riski vardır. Daha sonra çeşitli işlemlere ihtiyaç duyulur ve çocukluk
döneminde tekrarlı cerrahi işlemlerin yapılması gerekir. Eğik sırt
(skolyoz) korse ile tedavi edilebilir ancak bazen ameliyat gerekir. Eldeki
hatalar el cerrahı tarafından değerlendirilir ve ele alınır ve bazen el rayları
(ortezler) kullanılarak düzeltilebilir. Plastik cerrahi klinikleri ile
bağlantılı olarak, dudak, çene ve damak yarıkları olan çocukların nasıl takip
edileceği ve tedavi edildiği ile ilgili profesyonel ekipler vardır. Bu
ekipler plastik cerrah, konuşma terapisti, çene cerrahı, diş kontrol uzmanı ve
psikologu içerir. Rekonstrüktif cerrahi, burun büyümesi eksikliği ve yarık
damak için uygundur ve bu hastalar da konuşma terapisi
gerektirebilir. Solunum (solunum) problemleri ventilatör yardımı, özel
beslenme teknikleri ve göğüs fizik tedavisi dahil destekleyici tedavi
gerektirebilir. (2)
İşitme cihazını
düzenli olarak kontrol etmek ve gerekirse işitme bozukluğu ve işitme cihazları
için işitme uzmanıyla iletişim kurmak önemlidir. Bazı durumlarda koklear
implantlar (CI) kulağın arkasındaki derinin altındaki kafatası kemiğine
uygulanabilir. Koklear implant, ses sinyallerini doğrudan işitsel sinire
ileten bir verici türüdür. Bu yolla, tamamen sağır olan bir çocuk konuşma
ile iletişim kurmayı öğrenir. Çocuklar genellikle yaklaşık bir yaşında
ameliyat edilir. Larsen Sendromlu bireylerin tedavi süreçlerinin mutlaka uzman doktorlar
ve genetik danışmanlar tarafından takip edilmesi gereklidir. (2)
Trisomy 13, normal 2 kopya yerine,
vücut hücrelerinde 3 kopya kromozom 13’ün bulunmasıyla karakterize edilen bir
tür kromozom bozukluğudur. Etkilenen bazı insanlarda, hücrelerin sadece bir
kısmı ekstra kromozom 13’ü (mozaik trizomi 13 olarak adlandırılır) içerirken,
diğer hücreler normal kromozom çiftini içerir.Trisomy 13 ciddi zihinsel
sakatlığa ve doğuştan kalp defekti gibi birçok fiziksel anormalliklere neden
olur; beyin veya omurilik anormallikleri; çok küçük veya zayıf gelişmiş gözler
(mikroftalmi); ekstra parmak veya ayak parmakları; yarık damak veya damaksız
yarık dudak; ve zayıf kas tonusu (hipotoni). Vakaların çoğu kalıtsal değildir
ve sağlıklı ebeveynlerde yumurta veya sperm oluşumu sırasında rastgele bir
hatadan kaynaklanmaktadır. Hayatı tehdit edici çeşitli tıbbi problemler
nedeniyle, trizomi 13’ü olan birçok bebek, yaşamın ilk günleri veya haftaları
boyunca hayatta kalamaz.
Şekil.1. Patau Hastası Bir Birey (Yarık Dudak)
Şekil.2. Patau Hastası Bir Bireyin Eli (Ekstra Parmak)
Genetik
Değişiklikler ve Etken Faktörler
Trisomy 13 Sendromu olan
bireylerde, tümü veya nispeten büyük bir kromozom 13 bölgesi, hücrelerde iki
kez değil üç kez (trisomi) bulunur. Vakaların yaklaşık yüzde beşinde, sadece
yüzde bir hücre, ekstra 13. kromozomu (mozaikçilik) içerir. Kromozomlar tüm
vücut hücrelerinin çekirdeğinde bulunur. Her bireyin genetik özelliklerini
taşırlar. İnsan kromozom çiftleri, erkekler için eşit olmayan bir X ve Y
kromozomu çifti ve kadınlar için iki X kromozomu ile 1 ila 22 arasında
numaralandırılmıştır. Her kromozomun “p” olarak adlandırılan kısa bir kolu ve
“q” harfi ile tanımlanan uzun bir kolu vardır. Kromozomlar ayrıca
numaralandırılmış bantlara bölünmüştür. Belirli bir bölge veya kromozom 13
bölgelerinin tizomisi (veya “çoğaltma”), bozukluğu karakterize eden semptom ve
bulgulardan sorumludur. Semptomların şiddeti ve aralığı, kromozomun kopya
kısmının uzunluğuna ve konumuna bağlı olabilir. Ek olarak, trizomi 13
mozaikçiliği olanlar tipik olarak daha az ciddi semptomlara sahiptir; Bununla
birlikte, bu gibi durumlarda, hastalık belirtileri normalden neredeyse tüm
malformasyon spektrumuna kadar değişen değişkenlik gösterebilir. Trisomy 13
Sendromu olan çoğu kişide, kromozom 13’ün çoğalması, ebeveynlerden birinin
(örneğin mayoz bölünmesi sırasında yoksun olma) üreme hücrelerinin bölünmesi
sırasındaki spontan (de novo) hatalarından kaynaklanır. Kanıtlar, ileri yaşta
ebeveyn yaşı ile birlikte bu tür hataların riskinin artabileceğini
göstermektedir. Hücrelerin sadece bir yüzdesinin trizomi 13 anormallik
(mozaiklik) içerdiği durumlarda, döllenmeden sonra hücre bölünmesi sırasında da
hatalar oluşabilir (mitoz). Etkilenen bireylerin yaklaşık yüzde 20’sinde,
trizomi 13, kromozom 13 ve başka bir kromozom içeren bir translokasyondan
kaynaklanır. Translokasyonlar, belirli kromozom bölgeleri kopup yeniden
düzenlendiğinde meydana gelir, bu da genetik materyalin ve değiştirilmiş bir
kromozom setinin kaymasına neden olur.
Şekil.3. Patau Hastası Bir Bireyin Karyotip Analizi
Belirti ve
Semptomlar
Trisomy 13 Sendromu olan
bireylerde, ilişkili semptomların ve bulguların kapsamı ve ciddiyeti, kromozom
13’ün kopyalanmış (trisomik) kısmının spesifik konumuna ve ayrıca anormalliği
içeren hücrelerin yüzdesine bağlı olabilir. Bununla birlikte, etkilenen birçok
bebek ve çocukta, bu anormallikler gelişimsel gecikmeler, derin zihinsel
gerilik, olağandışı küçük gözler (mikroftalmi), üst dudaktaki anormal bir oluk
(yarık dudak), ağzın çatısının kapanmaması (yarık damak) olabilir. ), etkilenen
erkeklerde inmemiş testisler (kriptorşidizm) ve ekstra (süpernumerary) parmak
ve ayak parmakları (polidaktili). Baş ve yüz (kraniyofasiyal) alanın ek
malformasyonları da mevcut olabilir, örneğin eğimli bir alına sahip olan
nispeten küçük bir kafa (mikrosefali); geniş, düz bir burun; yaygın olarak
belirlenmiş gözler (oküler hipertelorizm); gözleri örten dikey cilt kıvrımları;
iç köşeler (epicanthal kıvrımları); kafa derisi kusurları; ve hatalı
biçimlendirilmiş, alçak ayarlanmış kulaklar. Etkilenen bebekler ayrıca beynin
belirli bölgelerinin (örneğin, ön beyin) eksik gelişimini de sağlayabilir;
böbrek (böbrek) malformasyonları; ve doğumda yapısal kalp (kalp) kusurları
(doğuştan). Örneğin, karakteristik kalp defektleri, kalbin üst veya alt
odalarını (atriyal veya ventriküler septal defektler) bölen bölümdeki anormal
bir açıklığı veya iki ana arter (aort, pulmoner arter) arasında ortaya çıkan
iki ana arter arasındaki fetal açıklığın sürekliliğini içerebilir kalp (patent
duktus arteriosus). Trisomy 13 Sendromlu birçok bebek beklenen oranda büyümez
ve kilo alamaz (gelişemez) ve şiddetli beslenme güçlüğü, azalan kas tonusu
(hipotoni) ve spontan beratlamanın (apnenin) geçici olarak kesildiği bölümler
vardır. Bebeklik döneminde veya erken çocukluk döneminde hayatı tehdit eden
komplikasyonlar gelişebilir.
Genetik Görülme
Sıklığı
Trisomy 13 Sendromu bazen sendromun
1960 yılında trizomik kökenini tanımlayan araştırmacılardan (Patau K) sonra,
Patau Sendromu olarak adlandırılır. Sendrom, kadınları erkeklerden biraz daha
sık etkiler ve yaklaşık 5.000 ila 12.000 canlı doğumda görülür. Kanıtlar,
tanınan tüm düşüklerin yaklaşık yüzde birinin, Trisomy 13 Sendromu ile birlikte
gerçekleştiğini göstermektedir. Ek olarak, yukarıda belirtildiği gibi, Trisomy
13’ün sıklığı annenin ilerleyen yaşı ile birlikte artar.
Kalıtım
Paterni/Deseni
Çoğu trizomi 13 vakası kalıtsal
değildir ve sağlıklı ebeveynlerde yumurta ve sperm oluşumu sırasında rastgele
olaylardan kaynaklanır. Hücre bölünmesinde bağlanma denilen bir hata, anormal
sayıda kromozom içeren üreme hücresi ile sonuçlanır. Örneğin, bir yumurta veya
sperm hücresi, ekstra bir kromozom 13 kopyası alabilir. Bu atipik üreme
hücrelerinin biri, bir çocuğun genetik yapısına katkıda bulunursa, çocuğun
vücudun her hücresinde ekstra bir kromozom 13 olacak.
Translokasyon trizomi 13 miras edilebilir. Etkilenmemiş bir kişi, 13 nolu
kromozom ile bir başka kromozom arasında genetik materyalin yeniden
düzenlenmesini taşıyabilir. Bu yeniden düzenlemelere dengeli translokasyonlar
denir, çünkü kromozom 13’ten fazla bir materyal yoktur. Kromozom 13 içeren
dengeli bir translokasyona sahip bir kişi, kromozom 13’ten çocuklarına ekstra
materyal geçirme şansını arttırır.
Teşhis Yöntemi ve
Tedavi
Bazı durumlarda, doğumdan önce
(doğum öncesi) fetal ultrasonografi, amniyosentez ve / veya koryon villus
örneklemesi (CVS) gibi özel testlerle Trisomy 13 Sendromu tanısı önerilebilir.
Fetal ultrasonografi sırasında, yansıyan ses dalgaları gelişen bir fetüsün
görüntüsünü oluşturarak, potansiyel olarak bir kromozomal bozukluk veya başka
anormallikler önerebilecek bulguları ortaya çıkarır. Trisomy 13 Sendromu tanısı
doğumdan sonra (doğum sonrası) ayrıntılı bir klinik değerlendirme,
karakteristik fiziksel bulguların tespiti ve kromozomal analiz ile konulabilir.
Testler ayrıca, embriyonik ve / veya fetal hemoglobinin Trisomy 13 Sendromlu
yenidoğan ve bebeklerin kanında olağandışı kalıcılığını ortaya çıkarabilir.
(Hemoglobin, kırmızı kan hücrelerinin oksijen taşıyan bileşenidir.) Sendromu
olan bebekler için, Trisomy 13 Sendromu ile potansiyel olarak ilişkili
koşulların erken tespitini ve uygun yönetimini sağlamak için dikkatli izleme ve
çeşitli özel testler yapılabilir. Trisomy
13 Sendromu’nun tedavisi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Böyle bir tedavi multidisipliner bir tıp uzmanları ekibinin koordine
çabalarını gerektirebilir. Bazı durumlarda, önerilen tedavi, hastalıkla
ilişkili bazı anormalliklerin cerrahi olarak düzeltilmesini içerebilir. Yapılan
cerrahi işlemler anatomik anormalliklerin doğasına ve ciddiyetine, bunlarla
ilişkili semptomlara ve diğer faktörlere bağlı olacaktır. Bu bozukluğu olan
çocuklar için destek ekibi yaklaşımı faydalı olabilir ve fizik tedavi, tıbbi ve
/ veya sosyal hizmetleri içerebilir. Genetik danışmanlık, Trisomy 13 Sendromu
olan çocukların aileleri için de faydalı olacaktır. Bu hastalığın diğer
tedavisi semptomatik ve destekleyicidir.
Genel Bilgi Polikistik böbrek hastalığı (PKD) , böbrekleri ve diğer organları etkileyen bir hastalıktır. Kist adı verilen sıvı dolu keseler kümeleri böbreklerde gelişir ve bu kistler böbreklerin kandan atık ürünleri filtreleme yeteneklerine müdahale eder. Kistlerin büyümesi böbreklerin genişlemesine neden olur ve böbrek yetmezliğine yol açabilir. Kistler ayrıca diğer organlarda, özellikle de karaciğerde gelişebilir.
Polikistik böbrek hastalığının sık görülen
komplikasyonları tehlikeli derecede yüksek kan basıncı ( hipertansiyon ), sırt veya yanlarda ağrı, idrarda kan (hematüri), tekrarlayan idrar
yolu enfeksiyonları, böbrek taşları ve kalp kapağı anormalliklerini içerir. Ek
olarak, polikistik böbrek hastalığı olan kişilerde anormal
şişkinlik ( anevrizma) riski daha yüksektir (aort denilen büyük bir kan damarı içinde veya beynin tabanındaki kan
damarlarında). Anevrizmalar yırtılırsa hayatı tehdit edici olabilir.
Polikistik böbrek hastalığının iki ana formu , normal başlangıç
yaşı ve aileden geçme şekli ile ayırt edilir. Otozomal dominant form
(bazen ADPKD olarak adlandırılır) tipik olarak yetişkinlikte başlayan belirti
ve semptomlara sahiptir, ancak böbreklerdeki kistler sıklıkla doğumdan veya
çocukluktan itibaren mevcuttur. Otozomal dominant polikistik böbrek
hastalığı , genetik nedene bağlı olarak tip 1 ve tip 2’ye
ayrılabilir. Polikistik böbrek hastalığının otozomal resesif
formu (bazen ARPKD olarak adlandırılır) daha nadirdir ve genellikle
yaşamın erken dönemlerinde ölümcüldür. Bu durumun belirti ve semptomları
genellikle doğumda veya erken bebeklik döneminde belirgindir.
Otozomal (veya İnfantil) resesif polikistik böbrek hastalığı (ARPKD),
böbreklerde sıvı dolu keseler (kistler) oluşumu ile karakterize nadir bir
genetik hastalıktır. Etkilenen bebeklerin çoğu yenidoğan (yenidoğan)
döneminde böbrekleri büyütmüştür ve bu sırada bazı vakalar ölümcül olabilir. ARPKD
basit bir böbrek hastalığı değildir ve vücudun ek organ sistemleri de özellikle
karaciğerde etkilenebilir. Yüksek tansiyon (hipertansiyon), aşırı
susuzluk, sık idrara çıkma ve beslenme güçlüğü de oluşabilir. Etkilenen
bazı çocuklar da belirgin yüz özelliklerine sahip olabilir ve solunum
yetersizliğine neden olan akciğerlerde (pulmoner hipoplazi) tam gelişmemiş
olabilir. Hastalığın ciddiyeti ve ortaya çıkan spesifik semptomlar bir
kişiden diğerine büyük ölçüde değişebilir. Etkilenen bazı çocuklar,
yaşamın ilk on yılında bir süre sonra son dönem böbrek yetmezliği
geliştirir. Bazı hastalarda, ergenliğe veya hatta yetişkinliğe kadar
semptomlar gelişmez.
Şu anda, PKD için bir tedavi yoktur. Ancak, semptomları kontrol altına
almak, kistlerin büyümesini yavaşlatmaya ve PKD’li insanlarda böbrek fonksiyon
kaybını önlemeye veya yavaşlatmaya yardımcı olmak için birçok destekleyici
tedavi uygulanabilir.
Hastalığı çocuklarına geçirmekten endişe duyan PKD’li bireyler, aile
planlaması konusunda kendilerine yardımcı olmak için bir genetik danışmanına
danışmak isteyebilirler.
Çoğu polikistik böbrek hastalığı vakasında otozomal dominant
kalıtım paterni vardır. Bu durumu olan insanlar , her hücrede bir PKD1 veya PKD2 geninin mutasyona uğramış bir kopyasıyla doğarlar . Bu
vakaların yaklaşık yüzde 90’ında, etkilenen bir kişi etkilenen bir ebeveynin mutasyonunu devralır. Vakaların diğer yüzde 10’u yeni bir mutasyondan
kaynaklanıyor Genlerin birinde ve ailesinde düzensizlik öyküsü olmayan kişilerde de
ortaya çıkar.
Her hücrede bir genin değiştirilmiş bir kopyası bozukluğa neden olmak için
yeterli olsa da , PKD1veya PKD2 geninin ikinci
kopyasındaki ek bir mutasyon kistlerin daha hızlı büyümesini sağlayabilir
ve hastalığın şiddetini artırabilir. Kistlerin genişleme ve böbrek
fonksiyon kaybına neden olma hızı geniş ölçüde değişir ve tanımlanmamış diğer
genlerdeki mutasyonlardan etkilenebilir.
Polikistik böbrek hastalığı da otozomal resesif paternde kalıtsal olabilir. Durumun bu formu olan insanlar , her hücrede PKHD1 geninin iki değiştirilmiş kopyası vardır . Otozomal
resesif bozukluğu olan bir çocuğun ebeveynleri etkilenmez fakat değiştirilmiş
genin bir kopyasının taşıyıcısıdır.
PKD1 , PKD2 , ve PKHD1 genleri mutasyonları polikistik böbrek hastalığına neden olur. PKD1 veya PKD2 genindeki mutasyonlar, otozomal
dominant polikistik böbrek hastalığına neden
olabilir ; PKD1 gen mutasyonları, ADPKD tip 1’e
ve PKD2 gen mutasyonları , ADPKD tip 2’ye neden
olmaktadır. Bu genler, fonksiyonları tam olarak anlaşılmayan proteinlerin
yapılması için talimatlar sağlar. Araştırmacılar, kimyasal sinyalleri hücrenin
dışından hücrenin çekirdeğine iletmekte yer aldıklarına inanıyor. İki
protein normal böbrek gelişimi, organizasyonu ve işlevini desteklemek için
birlikte
çalışır. PKD1 veya PKD2 genindeki mutasyonlar binlerce kistin oluşumuna yol açar, böbrek ve diğer organların normal fonksiyonlarını
bozan. PKD2 geninde mutasyon olan insanlar , özellikle
kadınlar, tipik olarak, PKD1 mutasyonları olan insanlardan daha
az şiddetli bir hastalık formuna sahiptir . Böbrek fonksiyonlarında
bir düşüş de dahil olmak üzere belirti ve semptomlar, yetişkinlerde daha sonra PKD2 mutasyonu olan
kişilerde ortaya çıkma eğilimindedir .
Mutasyonlar PKHD1 gen neden otozomal resesif polikistik
böbrek hastalığı . Bu gen, tam işlevi bilinmeyen bir proteini yapmak
için talimatlar sağlar; Bununla birlikte, protein muhtemelen hücrenin
dışından hücre çekirdeğine kimyasal sinyaller
iletir. Araştırmacılar, PKHD1 genindeki mutasyonların polikistik
böbrek hastalığının karakteristik sayısız kist oluşumuna neden
olduğunu belirlemedi .
Polikistik böbrek hastalığı genellikle genetik bir hastalık olmasına
rağmen , vakaların küçük bir yüzdesi gen mutasyonlarından
kaynaklanmamaktadır. Bu vakalara kazanılmış polikistik böbrek
hastalığı denir . Hastalığın bu şekli en çok birkaç yıl boyunca
hemodiyalizle tedavi edilen diğer böbrek hastalığı tipleri olan insanlarda
görülür (kandaki atık ürünleri filtreleyen bir prosedür).
Belirti ve Semptomlar
ARPKD’nin şiddeti ve ilerlemesi, aynı ailenin üyeleri arasında bile bir
kişiden diğerine büyük farklılıklar gösterebilir. Ağır vakalarda, ARPKD
bebeklik döneminde hayatı tehdit eden komplikasyonlara neden
olabilir. Diğer durumlarda, etkilenen bireyler, çocukluk veya ergenlik
döneminde daha sonraya kadar semptom geliştirmeyebilir.
Bazı çocuklar çocukluk döneminin başlarında böbrek nakli yapılmasına
ihtiyaç duyabilir; Diğerleri erken yetişkinliğe kadar nakil gerekmeyebilir
veya hiç olmayabilir. Nadir durumlarda, bireyler genç erişkinliğe kadar
semptom geliştirmeyebilir.
Genellikle, ilk başta hiçbir belirti yoktur. Daha sonra belirtiler
arasında;
Sırt ve alt taraflarda ağrı
Baş ağrısı
İdrarda kan
Genellikle, yaşamın ilerleyen dönemlerinde ARPKD gelişen kişilerde daha
hafif böbrek hastalıkları, fakat daha şiddetli karaciğer hastalığı olur.
ARPKD hakkındaki tıbbi literatürün çoğu, özellikle ARPKD hastalık geninin
tanımlanmasından önce yazılanlar, orantısız bir şekilde en ağır vakalara
odaklanmıştır. Bu nedenle, literatürün çoğu ARPKD’nin üniform olarak
ölümcül veya zayıflatıcı bir hastalık olduğu izlenimini verebilir. Araştırmacılar
artık ARPKD vakalarının hafif ila şiddetli olabileceğini biliyorlar. Sonuç
olarak, etkilenen bireylerin aşağıda belirtilen tüm belirtilere sahip
olmayacağını not etmek önemlidir. Etkilenen bireyler kendi özel
durumları, ilişkili semptomları ve genel prognozları hakkında doktorları ve
sağlık ekipleriyle konuşmalıdır.
ARPKD’nin karakteristik bulgusu böbreklerde sıvı dolu keseler (kistler)
gelişmesidir. Etkilenen tüm bireyler böbreklerde kist gelişir, ancak kist
gelişiminin sayısı, büyüklüğü, ilerlemesi ve ciddiyeti bir kişiden diğerine
büyük farklılıklar gösterir. Etkilenen bireylerin çoğunda, böbrek kistleri
uteroda büyür ve çoğalır, böbreklerde anormal genişlemeye neden
olur. Büyümüş böbrekler doğumda veya yenidoğan döneminde belirgin olabilir. Bu
bebeklerde böbrekler sert ve her iki yanda da hissedilir (elle
hissedilebilir). Kistik böbreklerle ilişkili ek semptomlar arasında yüksek
tansiyon (hipertansiyon) ve yan ağrısı vardır. ARPKD’li çocuklarda yüksek
tansiyon yaygındır ve yaygın, şiddetli ve yönetimi zor olabilir.
Ağır ARPKD vakalarında, etkilenen bebekler doğumdan kısa bir süre sonra,
özellikle solunum (solunum) yetersizliği veya yetersizliği ile hayatı tehdit
eden komplikasyonlarla karşılaşabilir. Solunum güçlüğü genellikle
hamilelik sırasındaki yetersiz amniyotik sıvı seviyesinden (oligohidramnios)
kaynaklanmaktadır. Akciğerlerin doğru gelişimini önleyen kitlesel
genişlemiş böbrekler, solunum yetmezliği veya yetersizliğine de katkıda
bulunabilir. Her ne kadar bazı çocuklar yenidoğan döneminde hayatta
kalmasalar da (tahminlerin çoğuna göre yaklaşık yüzde 30), çocukların çoğunluğu
yaşayabilir.
Yenidoğan döneminin ötesinde hayatta kalan çocuklar, böbreklerin
boyutlarını küçültebilse de, genellikle kötüleşen böbrek fonksiyonlarını
geliştirirler. Çocukların çoğu, geç çocukluk, ergenlik veya genç
erişkinliğe kadar kronik böbrek yetmezliği geliştirmez. Böbrek yetmezliği,
böbreklerin temel işlevlerini yerine getirme yeteneğinin azalması anlamına
gelir. Böbrekler, göğüs kafesinin altına yerleştirilmiş iki fasulye
şeklinde organdır. Böbrekler, atık ürünlerin kandan ve vücuttan süzülmesi
ve atılması, belirli hormonların yaratılması ve vücuttaki potasyum, sodyum,
klorür, kalsiyum ve diğer elektrolitler gibi bazı kimyasalların dengesinin
korunmasına yardımcı olmak gibi çeşitli fonksiyonlara sahiptir. ARPKD’de
böbreklere verilen hasar yavaşça ilerleyici olabilir ve halsizlik, iştahta
değişiklikler, şişlik, sırt ağrısı gibi çeşitli semptomlara neden
olabilir, zayıf sindirim, aşırı susama ve sık idrara çıkma. Sonunda,
birçok çocuk böbrek hastalığına son aşamada ilerler.
Şiddetli ARPKD vakalarında, etkilenen bebeklerde aşırı derecede büyümüş böbrek
vardır ve doğumda idrar üretimini azaltır. Uterodaki azalmış idrar
üretimi, gelişen bir fetüsü çevreleyen sıvı olan amniyon sıvısının
(oligohidramnios) eksikliğine katkıda bulunur. Bir fetusun korunmasına ve
desteklenmesine ek olarak, amniyotik sıvı büyüme faktörlerini ve fetal gelişim
için hayati olan diğer maddeleri içerir. Düşük amniyotik sıvı seviyeleri
akciğer gelişimini bozabilir ve sonuç olarak, etkilenen bazı bebeklerde az
gelişmiş akciğerler (pulmoner hipoplazi) olabilir. Bu bebekler yenidoğan
döneminde ciddi, hayatı tehdit edici solunum (solunum) komplikasyonları
yaşayabilir. Oligohidramniyoslu bebekler ayrıca derin gözler, düz bir
burun, küçük bir çene (mikrognati) ve anormal, düşük ayarlı kulaklar gibi
belirgin yüz özellikleri de geliştirebilir.
Böbreklere ek olarak karaciğer, ARPKD’li çocuklarda ve yetişkinlerde de
yaygın olarak etkilenir. Karaciğer vücutta gıdaları enerjiye ve besin
maddelerine dönüştürmek, vitamin depolamak ve vücuttaki toksinleri süzmek dahil
olmak üzere birçok işlevi yerine getirir. ARPKD’li çocuklar, aşırı lif
benzeri bağ dokusunun karaciğere yayıldığı konjenital hepatik fibrozis olarak
bilinen bir karaciğer hastalığı geliştirir. Her ne kadar çocuklarda
konjenital hepatik fibrozis olsa da, tüm çocuklar karaciğer fonksiyon bozukluğu
geliştirmez. ARPKD’de ortaya çıkabilecek karaciğer anomalileri,
karaciğerin genişlemesini (hepatomegali), karaciğerden safra taşıyan tüplerin
(safra kanallarının) iltihaplanması ve enfeksiyonunu ve safra kesesi ve
bağırsaklara (kolanjit) ana damarının yüksek kan basıncını içerir. karaciğer
(portal hipertansiyon).
Portal hipertansiyon, yemek borusu, mide ve bağırsakların damarlarında
(varislerde) artan baskı ve şişmeye (dikkat) neden olabilir. Bu damarlar
yırtılabilir ve potansiyel olarak hayati tehlike yaratan gastrointestinal
kanamaya (varis kanaması) neden olabilir. Etkilenen çocuklar ilerleyici
karaciğer fonksiyon bozukluğu ve yara izi (siroz) ve nihayetinde karaciğer
yetmezliği yaşayabilir.
ARPKD’li çocuklar, yetersiz böbrek fonksiyonu veya midenin genişlemiş
böbrekler, karaciğer ve / veya dalakla sıkışması nedeniyle beslenme güçlüğü
çekebilir. Beslenme zorlukları ve kronik böbrek yetmezliği, etkilenen
bireylerde zayıf büyümeye katkıda bulunabilir. Bazı çocuklar idrar yolu
enfeksiyonlarına yatkın olabilir ve su ve tuz dengesi ile ilgili problemleri
olabilir.
ARPKD’li bazı çocuklar dalağın genişlemesini yaşayabilir
(splenomegali). Splenomegali potansiyel olarak düşük seviyelerde kırmızı
kan hücreleri (anemi), trombositler (trombositopeni) ve beyaz kan hücreleri
(lökopeni) ile sonuçlanabilir. Anemi yorgunluk, soluk cilt, düzensiz kalp
atışı ve nefes darlığına neden olabilir. Trombositopeni kolay morarma,
kesiklerden uzun süre kanama, spontan burun kanaması ve ciltte yüzeysel kanama
ile sonuçlanabilir (peteşiler). Lökopeni vücudun enfeksiyon ve
hastalıklarla savaşma yeteneğini azaltır.
Genetik Görülme Sıklığı Polikistik böbrek hastalığı oldukça yaygın bir genetik hastalıktır. Amerika Birleşik Devletleri’nde yaklaşık 500.000 kişiyi etkiliyor. Hastalığın otozomal dominant formu otozomal resesif formdan çok daha yaygındır. Otozomal dominant polikistik böbrek hastalığı 500 ila 1.000 kişiden 1’ini etkilerken, otozomal resesif tip 20.000 ila 40.000 kişiden 1’inde görülür.
Hastaların çoğuna utero veya doğumda tanı konmasına rağmen, hafif vakalar
ergenliğe veya yetişkinliğe kadar belirgin olmayabilir.
Teşhis Yöntemleri ve Tedavileri
Nedenleri
ARPKD, otozomal resesif bir özellik olarak kalıtılan PKHD1 geninin mutasyonlarından
kaynaklanır . Genetik hastalıklar, baba ve anneden alınan
kromozomlarda bulunan belirli bir gen için alellerin kombinasyonu ile
belirlenir.
Resesif genetik bozukluklar, bir birey hastalık geninin anormal bir
kopyasını her bir ebeveynden aldığında ortaya çıkar. Bir birey genin
normal bir kopyasını ve bir mutasyonu olan birini alırsa, kişi hastalık için
taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki taşıyıcı
ebeveynin hem kusurlu geni geçmesi hem de etkilenen bir çocuğa sahip olma riski
her hamilelikte yüzde 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi
olma riski her hamilelikte yüzde 50’dir. Bir çocuğun her iki ebeveynden
normal gen alma ve bu özellik için genetik olarak normal olma şansı yüzde
25’tir. Risk erkeklerde ve kadınlarda aynıdır.
Araştırmacılar çoğu tipik ARPKD vakasının tek bir gene, özellikle de PKHD1genine mutasyondan
kaynaklandığını belirlemiştir . PKHD1 geni kromozom 6
(6q21.1) uzun kolu (q) üzerinde yer almaktadır. PKHD1 büyük
bir gendir ve bu gene birçok farklı mutasyon ARPKD’ye neden olur.
Otozomal dominant polikistik böbrek hastalığı (ADPKD), böbreklerde kist
oluşumu ile karakterize genetik bir hastalıktır. ADPKD dominant bir
hastalık olduğundan, etkilenen bireylerin çoğunda ARPKD’den farklı olarak aile
PKD öyküsü vardır. Böbreklerdeki kist oluşumunun neden olduğu semptomlar
arasında yüksek tansiyon (hipertansiyon), son kaburga ile kalça arasında
vücudun yanlarında ağrı (gögüs ağrısı), idrarda kan (hematüri) ve böbreklerin
aşamalı olarak zayıf fonksiyonu bulunur ( böbrek yetmezliği). Hastaların
en az yüzde 50’sinde ADPKD, sonunda diyaliz veya böbrek nakli olmak üzere renal
replasman tedavisi gerektiren evre böbrek hastalığına son verir. ADPKD
basit bir böbrek hastalığı değildir ve vücudun diğer organ sistemleri kistlerin
ve diğer hastalıkların gelişmesinden potansiyel olarak etkilenebilir (çoklu
sistem bozukluğu). Her insanda bulunan spesifik semptomlar, ilgili
spesifik organ sistemlerine bağlıdır. Karaciğer, pankreas, omuriliği ve
beyni kaplayan bir zar (araknoid membran) ve semen (seminal veziküller) parçası
olan sıvı üreten erkek üreme sisteminin bezleri kistler
gelişebilir. ADPKD’li bireylerde kalbi ve kan damarlarını (kardiyovasküler
sistem) etkileyen anormallikler de görülebilir.
Çeşitli farklı bozuklukların bir özellik olarak kistik böbrekleri
olabilir. Bu bozukluklara bazen polikistik böbreğin sendromik biçimleri de
denebilir. Bu bozukluklar genellikle ek nörolojik, dijital, göz ve / veya
diğer semptomlara ve bunları ARPKD’den ayıran fiziksel bulgulara
sahiptir. Bu bozukluklar arasında Bardet-Biedl sendromu, Meckel sendromu,
Joubert sendromu, Senior-Loken sendromu, nefronofthis ve oral-fasiyal-dijital
sendrom bulunur.
Teşhis
ARPKD, doğumdan önce klinik bulgulara dayanarak şüphelenebilir (örneğin,
elle hissedilen göbek kitlesi, az gelişmiş akciğerler, oligohidramnios ve
hipertansiyon). ARPKD teşhisi için sonogram, ultrason ve manyetik rezonans
görüntüleme (MRG) içeren radyolojik görüntüleme kullanılabilir. Böbrek
anormalliklerinin tespitine ek olarak, karaciğerdeki intraheptik kanalların
tıkayıcı olmayan genişlemesini (dilatasyonu) tanımlamak için çeşitli radyolojik
görüntüleme teknikleri de kullanılabilir.
Prenatal ultrason, genişlemiş böbrekleri ortaya çıkarabilir (bazı
durumlarda doğumdan sonraki 18 hafta gibi). Bir ultrason ayrıca, gerici
toplama tüpleri olan sayısız küçük kisti de açığa çıkarabilir. Gerçek
böbrek kistleri de mevcut olabilir. Bununla birlikte, bir ultrason böbrek
büyümesini veya oligohidramniosu tespit etmede başarısız olabilir.
PKHD1 genlerindeki mutasyonlar için genetik testler birçok farklı
laboratuarda mevcuttur.
ARPKD tanısı konmuş en az bir hamilelik geçirmiş risk altındaki bazı
aileler için doğum öncesi veya doğum öncesi genetik tanı için genetik testler
kullanılabilir.
Tedavi
ARPKD’nin tedavisi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Özel tedaviler, böbrek ve karaciğer fonksiyonunun korunmasına
yöneliktir. Bebeklik döneminde solunum güçlüğü çeken birçok çocuk nefes almanıza
yardımcı olmak için mekanik ventilasyon gerektirebilir. Nitrik oksit gibi
ilaçlar, akciğerlere oksijen vermek (oksijenlemek) için yardımcı olabilir.
Şiddetli vakalarda, idrar üretimindeki (oligurya) azalmış veya idrar geçişi
(anüri yok) yaşayan yenidoğanlarda yaşamın ilk birkaç günü periton diyalizi
gerekebilir.
İlaçlar yüksek kan basıncını, özellikle de anjiyotensin dönüştürücü enzim
(ACE) inhibitörlerini kontrol etmek ve yönetmek için kullanılabilir. Bazı
kişilerde, yüksek tansiyon tedaviye dirençli olabilir (refrakter) ve birden
fazla ilaç gerektirecek kadar şiddetli olabilir. Antibiyotikler idrar yolu
enfeksiyonları veya kolanjit tedavisinde kullanılabilir.
Bazı çocuklar D vitamini, demir, bikarbonat ve sitrat gibi besin
takviyelerine ihtiyaç duyabilir. Yeterli sıvı ve tuz takviyesi de gerekli
olabilir. Beslenme zorlukları ve büyüme gecikmeleri nedeniyle, bazı
çocuklar midede küçük bir cerrahi açıklıktan (gastrostomi) bir tüp veya burun
içinden, özofagustan aşağı ve mideye (nazogastrik tüp) tüp yerleştirmeyi
gerektirebilir. Bu tüpler temel besinleri doğrudan sağlamak için
kullanılır. Ciddi durumlarda, büyüme hormonu tedavisi gerekli olabilir.
Böbreklerin artık çalışmadığı son dönem böbrek hastalığı olan kişiler,
diyaliz veya böbrek nakli gerektirir. Diyaliz, bir makinenin, böbrek –
atık ürünlerin kan dolaşımından süzülmesini, kan basıncını kontrol etmeyi ve
potasyum gibi gerekli temel kimyasal seviyelerini korumaya yardım etmesini
sağlamak için kullanılan bir prosedürdür. Son dönem böbrek hastalığı geri
dönüşümsüz değildir, bu nedenle bireyler yaşam boyu diyaliz tedavisi veya
böbrek nakli gerektirecektir. Böbrek fonksiyon bozukluğunun son dönem
böbrek hastalığına ilerleme hızı, bir kişiden diğerine büyük ölçüde
değişebilir. Bazı bireyler çocukluk döneminde böbrek nakli
gerektirir; diğerleri yetişkinliğe kadar nakil gerektirmeyebilir veya hiç
olmayabilir.
Progresif portal hipertansiyon, portal ven ile inferior vena cava arasında,
vücudun kanının üçte ikisinden kan akıtan ana ven arasında bir bağlantı yapılan
portakaval şantla tedavi gerektirebilir. Portal venin yüksek kan basıncını
azaltmak için portacaval şant tasarlanmıştır.
Varis kanaması tıbbi bir acil durumdur ve acil tedavi
gerektirir. Varis kanaması, sodyum klorid gibi bir çözeltinin etkilenen
bir kan damarı içine enjekte edildiği bir prosedür olan skleroterapi ile tedavi
edilebilir. Solüsyon kan damarını tahriş eder ve sonunda kanın
pıhtılaşmasına neden olur. ARPKD’li bireylerin küçük bir yüzdesi sonunda
karaciğer nakli gerektirebilir.
Erythropoietin, ARPKD’li bazı çocuklarda anemi yaşayan kırmızı kemik
hücreleri üretmek için kemik iliğini stimüle etmek için
kullanılabilir. Dalakların cerrahi olarak çıkarılması (splenektomi) bazı
durumlarda şiddetli splenomegali tedavisinde kullanılır.
Genetik danışma, etkilenen bireyler ve aileleri için faydalı
olabilir. Diğer tedavi semptomatik ve destekleyicidir.
Mesane veya böbrek enfeksiyonları. Böbrek hasarını önlemek için
antibiyotik ile enfeksiyonların derhal tedavisi gereklidir.
PKD, tansiyonu kontrol etmek için yediklerinizde değişiklik
gerektirebilir. Sağlıklı bir beslenme planının ardından kan basıncını
düşürmeye yardımcı olabilir.
Doktorunuzun önerdiği Sağlıklı bir kiloyu koruyun. Sigara içilmemeli,
içiliyorsa bırakılmalı.
Düzenli egzersiz. Haftanın çoğu günü en az 30 dakika orta derecede
fiziksel aktivite,
Alkol kullanımını sınırlandırılmalı, gün boyunca bol miktarda sade su içmek,
tüm içeceklerde kafeinden kaçınılması öneriler arasındadır.
Nisan 2018’de FDA, otozomal dominant polikistik böbrek hastalığının (ADPKD)
tedavisi için “tolvaptan” adlı yeni bir ilacı onayladı. İlaç, bu tip PKD
için risk altında olan yetişkinlerde böbrek fonksiyonlarının yavaşlamasını
sağlamak için kullanılabilir.
Diğer kronik hastalıklarda olduğu gibi polikistik böbrek hastalığına sahip
olmak bunaltıcı gelebilir. Arkadaşların ve ailenin desteği, kronik bir
hastalıkla baş etmede önemlidir. Ek olarak, bir danışman, psikolog,
psikiyatrist veya din adamı üye yardımcı olabilir.
Hastalığın Diğer İsimleri
Polikistik Böbrek Hastalığı
(PKD)
Otozomal resesif polikistik
böbrek hastalığı (ARPKD)
Otozomal dominant polikistik
böbrek hastalığı (ADPKD)
Von
Willebrand hastalığı, kan pıhtılaşma sürecini yavaşlatan bir kanama bozukluğu
hastalığıdır. Bu hastalığa sahip bireylerde burun kanaması, morarma, ameliyat
veya diş çekilme sonrası uzun süreli kanama görülebilir. Kadınlarda adet
kanaması uzun süreli olabilir. Hastalığın çok ilerlemiş durumlarında, küçük
yaralanmalarla birlikte veya yaralanma olmasa bile şiddetli kanama görülebilir.
Bu hastalığın üç tipi vardır: Tip 1, Tip 2, Tip 3. Tip 1 en hafif ve en çok
görülen, Tip 3 ise hastalığın en şiddetli ve nadir görülen halidir.
Genetik Değişiklikler/Etken Faktörler
VWF genindeki
mutasyonlar Vol Willebrand hastalığına sebep olur. VWF geni, kan pıhtılaşmasını
sağlayan proteinin (Von Willebrand faktörü) üretilmesinde rol oynar. Bu protein
kan pıhtılaşması ve bir yaralanmadan sonra yaşanacak kan kaybını önlemek için
önemlidir. Eğer Von Willebrand faktörünün fonksiyonu bozulursa, kan pıhtıları
düzgün bir şekilde oluşamazlar. VWF genindeki mutasyonlar bu proteinin
fonksiyonunu bozar ve iyi çalışmasını önler.
Belirti ve Semptomlar
-Epistaksis (burun kanaması): sık sık ve kanamanın zor durması
-Vücutta bir yerin kolay morarması: küçük yaralanmalarda bile büyük morarmalar
Aort kapağının daralması -Diş operasyonundan sonra diş etinde uzun süreli kanama
Kadınların adet döneminde uzun süreli veya şiddetli kanama
İdrarda kan görülmesi -Ameliyat sonrası şiddetli kanama
-Küçük yaralanmalarda şiddetli kanama
Genetik Görülme Sıklığı
Von Willebrand hastalığı,
erkek ve kadınları eşit oranda etkilemektedir ama kadınlarda adet dönemindeki
şiddetli kanamadan dolayı teşhisi daha kolay olmaktadır. Herhangi bir yaşta teşhis
edilebilir. 1.000.000 kişide
23-110 kişi bu hastalığa sahip olduğu tahmin ediliyor. VWD tip 3 en nadir görülendir.
Kalıtım Paterni/Deseni
Tip 1 ve tip
2A ayrıca tip 2B ve 2M otozomal dominant olarak kalıtılmaktadır. Tip 2N ve tip
3 ve bazı tip 1 ve tip 2A otozomal resesif olarak kalıtılmaktadır.
Teşhis Yöntemleri ve Tedaviler
Tip 1 ve tip 2 hastalarının teşhisi daha zordur ve genelde ameliyat sonrası şiddetli kanamada belli olur. Tip 3’ün teşhisi daha kolaydır çünkü kan pıhtılaşması küçük yaralanmalarda bile çok yavaştır. Teşhis için koagülasyon testleri (Screening coagulation test) yapılabilir ve böylece ne kadar sürede kanın pıhtılaştığı anlaşılabilir. Tedavi her tip için farklılık gösterebilir ve bu ilaçlar tedavi için kullanılabilir: Antihemophilic factor (human) (Markası: Alphanate®), Antihemophilic factor/von Willebrand factor complex (human), dried, pasteurized (Markası: Humate-P), Desmopressin acetate (Markası: Stimate), Recombinant von Willebrand factor (rhVWF) (Markası: Vonvendi), Human Plasma Coagulation Factor VIII And Human Plasma Von Willebrand Factor (Markası: Wilate(R)).
Başka bir tedavi de desmopressin hormonu verilmesidir. Genelde burun spreyi veya enjeksiyon ile verilir. Bu hormon Von Willebrand faktörünün üretimini artırır. Bu tedavi genelde tip 1 ve tip 2 için daha çok işe yarar.
Okülo-aurikülo-vertebral
spektrum (OAVS), birçok klinisyenin birbiriyle yakından ilişkili olduğuna
inandığı ve aynı hastalığın ciddiyet aralığını temsil eden üç nadir hastalığa
karşılık gelir. Bu bozukluklar doğumda belirgindir
(doğuştan). Adından da anlaşılacağı gibi, gözler, kulaklar ve omurganın
malformasyonlarını içerir.
Okülo-aurikülo-vertebral
bozukluk (OAVD) hastalığın en hafif formunu temsil ederken, Goldenhar sendromu
en şiddetli form olarak ortaya çıkar. Hemifakiyal mikrostomi orta bir form
gibi görünmektedir.
Bozukluk,
durumdan duruma değişiklik gösterebilen çok çeşitli belirtiler ve fiziksel
özellikler ile tanımlanır. Bununla birlikte, bu tür anormallikler elmacık
kemikleri, çene, ağız, kulaklar, gözler ve / veya omurganın (omurlar)
kemiklerini tutma eğilimindedir. Çoğu durumda (yaklaşık% 60), bu tür
malformasyonlar vücudun bir tarafını (tek taraflı) etkilese de, etkilenen
bireylerin yaklaşık yüzde 10 ila 33’ü vücudun her iki tarafında bu tür
malformasyonlara sahiptir (iki taraf), bir taraf tipik olarak daha fazla
etkilenir diğerinden (asimetri). Bu gibi vakaların çoğunda, sağ taraf soldan
daha ciddi bir şekilde etkilenir.
Çoğu durumda, OAVS rastlantısal olarak ortaya çıkar, görünür bir nedeni yoktur (sporadik). Bununla birlikte, bazı durumlarda, aile öyküleri otozomal dominant veya resesif kalıtım gösterir. Ek olarak, bazı araştırmacılar bu bozukluğa, muhtemelen çevresel faktörlerle (çok faktörlü kalıtım) kombinasyon halinde birçok genin etkileşiminden kaynaklanabileceğini öne sürmektedir.
Çoğu
durumda, okülo-aurikülo-vertebral spektrum belirgin bir neden olmadan
(sporadik) rastgele ortaya çıkar. Bununla birlikte, bazı durumlarda,
otozomal dominant veya daha az sıklıkla otozomal resesif kalıtım öneren pozitif
aile öyküleri tespit edilmiştir. Ek olarak, birçok araştırmacı, OAVS’nin,
muhtemelen çevresel faktörlerle (çok faktörlü kalıtım) kombinasyon halinde
birçok genin etkileşiminden kaynaklanabileceğini öne sürmektedir.
Açıklanamayan
nedenlerden dolayı, hamilelik sırasında belirli ilaçlara (örneğin, retinoik
asitli bazı akne ilaçları) ya da koşullara (örneğin diyabet) maruz kalmış
kadınların, OAVS’ın özellik anormallikleri olan çocuklar olduğu
anlaşılmaktadır. Ek olarak, OAVS ile ilgili ayırt edici özellikler de
çeşitli kromozomal bozukluklarla birlikte ortaya çıkmıştır.
Belirti ve Semptomlar
Okülo-aurikülo-vertebral
spektrum doğumda belirgin olan üç nadir bozukluğu temsil eder (konjenital) ve
vakadan vakaya büyük ölçüde değişiklik gösterebilen geniş bir semptom yelpazesi
ve fiziksel özellikler ile karakterize edilir. Bununla birlikte, bu tür
anormallikler elmacık kemiklerini, çeneleri, ağzı, kulakları, gözleri ve / veya
omurganın (omur) kemiklerini tutma eğilimindedir. Vakaların yaklaşık yüzde
60’ında, bu tür malformasyonlar vücudun bir tarafını (tek taraflı)
içerir. Ancak, etkilenen bireylerin yaklaşık yüzde 10 ila 33’ünde, vücudun
her iki tarafı da tutulabilir (iki taraflı), bir taraf diğerinden daha fazla
etkilenir (asimetri). Bu gibi birçok durumda, sağ taraf soldan daha ciddi
şekilde etkilenir.
Bilinmeyen nedenlerden
dolayı hemifasiyal mikrozomi (HFM) yüzün sadece sağ tarafını etkileme
eğilimindedir. HFM’de, hem çene hem de göz etkilenen tarafta büyük ölçüde
daha küçük olabilir. Etkilenen taraftaki yanak, o taraftaki elmacık
kemiklerinin gelişmesi nedeniyle daha düz görünebilir. Dış kulak daha
küçük (mikrotia) veya hatta yok (anotia) olabilir. İşitme kaybı da
olabilir. İstihbarat etkilenmez.
OAVS’nin Goldenhar
varyantına sahip kişiler, çoğu HFM belirtisinin tümü olmasa da ortaya çıkar,
ancak vakaların yüzde 10 ila 33’ünde, semptomlar yüzün her iki tarafını da
etkiler (iki taraflı). Yarık dudak ve / veya yarık damak mevcut olabilir,
ancak tek başına yarık damak varlığı daha yaygındır. Dilin ve yanakların
kasları konuşmada ciddi zorluklara neden olabilir. Gözün bazı dokuları
kapanmayabilir ve değişken büyüklükte bir çentik (koloboma) şeklinde ortaya
çıkabilir. Vakaların yaklaşık üçte birinde, hasta gözde bir kist (dermoid
kist) ile kendini gösterir. Ayrıca, Goldenhar sendromlu hastalar, kalp
defekti ve böbrek problemleri ile birlikte ortaya çıkabilir. Goldenhar
sendromlu kişiler bir tarafta az gelişmiş böbreklere ve hatta etkilenen tarafta
böbrek eksikliğine sahip olabilir. İki veya daha fazla omur birbirine
kaynaşmış veya örülmüş olabilir.
Genetik Görülme Sıklığı
OAVS, erkekleri
kadınlardan yaklaşık 3: 2 oranında daha sık etkiler. Tıp literatüründe
bozukluğun ortaya çıkma oranı ile ilgili bazı anlaşmazlıklar vardır. Rapor
edilen tahminler 3000 ila 5000 canlı doğumdan birinden, 25.000-40.000 canlı
doğumdan birine kadar değişmektedir. OAVS ile ilişkili fiziksel
özelliklerin çoğu, doğumda (doğuştan) belirgindir; çoğu durumda yaklaşık dört
yaşına kadar belirgin olmayabilecek olası yüz asimetrisi dışındadır.
Kalıtım Paterni /Deseni
Okülo-aurikülo-vertebral
spektrum (OMIM164210) kulağın anomalileri (çoğunlukla mikrotia), hemifasiyal
mikrozomi ve vertebral kolonun defektleri ile karakterize fenotipik ve
muhtemelen genetik olarak heterojen bir hastalıktır. İlişkili klinik
bulgular göz ve beyin anomalilerini ve gelişimsel gecikmeyi içerir.
Kraniyofasiyal Mikrozomi Genel Bakış: Dahil Olan Fenotipler
Hemifakiyal mikrozomi Okülo-aurikülo-vertebral spektrum Goldenhar sendromu Birinci ve ikinci branşsal ark sendromu Otomandibular disostoz Facio-auriculo-vertebral sendrom Lateral yüz displazisi
Kraniyofasiyal
mikrozomi (CFM), öncelikle birinci ve ikinci dal kemerlerinden türetilen
yapıları içeren bir tür yanlış biçimlendirme spektrumunu
içerir. Karakteristik bulgular maksiller ve / veya mandibular hipoplaziden
kaynaklanan yüz asimetrisini; preauriküler veya yüz etiketleri; mikrotiya
(dış kulağın hipoplazisi), anotia (dış kulak yokluğu) veya aural atreziyi (dış
kulak kanalının yokluğu) içerebilen kulak malformasyonları; ve işitme
kaybı. Ciddiyet, normal görünen bir kulağın önünde küçük bir cilt etiketi
bulunan ince yüz asimetrisinden, iki taraflı tutuluma (tipik olarak asimetrik),
kulak kanallarının atrezili mikrorotiyal / anotiye, mikroftalmi ve ağır
mandibular hipoplaziden solunum yetmezliğine kadar değişebilir. Yarık
dudak ve / veya damak dahil diğer kraniyofasiyal malformasyonlar görülebilir.
CFM en
sık bilinmeyen etiyolojiye sahip tek taraflı bir vaka (yani, ailedeki tek bir
bireyde ortaya çıkma) olarak ortaya çıkar ; tekrarlama riskleri
ampiriktir. CFM’li bir kişinin kalıtsal veya de novokromozom anomalisine sahip olduğu
tespit edilirse , bu durum için genetik danışma belirtilir. Bazen otozomal
dominant veya otozomal resesif kalıtım
görülür. Bir proband CFM’ye sahipse
ve CFM ailesinde rapor edilmemiş bir aile öyküsü yoksa, sibs riski% 2 -% 3’dür,
ancak bu düşük penetrasyon ve
bazı ince özellikler için doğru aile öyküsü elde etmenin zorluğu nedeniyle
hafife alınabilir .
Teşhis Yöntemleri ve Tedavileri
Teşhis
Nadiren,
okülo-aurikülo-vertebral spektrum doğumdan önce (doğum öncesi) ultrason
görüntüleme gibi özel testlerle tespit edilebilir. Fetal ultrasonografide,
yansıyan ses dalgaları, gelişmekte olan fetüsün bir görüntüsünü oluşturmak için
karakteristik bulguları ortaya çıkarmak için kullanılabilir. OAVS
durumunda, bu bulgular alt çenede (mandibula) kemik varlığına veya yokluğuna,
dış kulaklarda ciddi anormalliklere, yarık damakta ve / veya yarık dudağa
bağlıdır.
OAVS ayrıca doğumdan
sonra (doğum sonrası) ayrıntılı bir klinik değerlendirme, karakteristik
fiziksel bulguların belirlenmesi ve ileri görüntüleme teknikleri ile teşhis
edilebilir ve / veya doğrulanabilir.
Potansiyel olarak
okulo-aurikülo-vertebral spektrum bozuklukları ile ilişkili spesifik
anormallikleri doğrulamak için çeşitli uzmanlık testleri
yapılabilir. Örneğin, bilgisayar destekli tomografi (BT) taraması, işitme
kaybına katkıda bulunabilecek orta kulak anormalliklerinin tespitinde önemli
bir yardımcı olabilir. Gelişmiş görüntüleme teknikleri ayrıca kafatasının,
omurganın, akciğerlerin ve / veya böbreklerin diğer potansiyel anormalliklerini
tespit etmek ve / veya onaylamakta yardımcı olabilir. Bazı durumlarda,
hastalıkla ilişkili olabilecek konjenital kalp kusurlarının varlığını tespit
etmek ve / veya doğrulamak için ek özel testler (örneğin, ekokardiyogramlar,
elektrokardiyogramlar, kalp kateterizasyonu, özel röntgen çalışmaları vb.) yapılabilir.
.
Mikroftalmi veya
anoftalmi, epibulbar dermoidler ve lipodermoidler, şaşılık, vb. Gibi bazı göz
(oküler) anormallikleri tespit etmek, doğrulamak ve / veya karakterize etmek
için gözün içini görselleştiren bir aletle (opthalmoscope) inceleme
yapılabilir.
OAVS’li yenidoğanlarda
yutma ve beslenme zorlukları, özofagus atrezisi ve trakeoözofageal fistül gibi
anormallikler gösterebilir. Bu anormallikler, sıvıyı vücuda enjekte etmek
veya sıvıyı vücuttan boşaltmak için kullanılan esnek, içi boş bir tüp
aracılığıyla tespit edilebilir (kateter). Ağızdan mideye geçemezse
konjenital malformasyonlar mevcut olabilir.
Tedavi
OAVS
tedavisi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Tedavi, tedaviye kapsamlı ve sistematik bir yaklaşım sağlamak
için birlikte çalışması gerekebilecek bir uzmanlar ekibinin koordine çabalarını
gerektirebilir. Bu uzmanlar çocuk doktorlarını içerebilir; kulak,
burun ve boğaz rahatsızlıklarını teşhis ve tedavi eden doktorlar (kulak burun
boğaz uzmanları); göz uzmanları (göz doktorları); nöroloji; kalp
(kardiyologlar) ve / veya akciğer (kardiyotorasik) cerrahları; böbrek
(nefrologlar), idrar yolu (ürologlar) ve sindirim sistemi (gastroenterologlar)
rahatsızlıklarının tanı ve tedavisinde uzmanlaşmış doktorlar; estetik
cerrahlar; işitme problemlerini değerlendiren ve tedavi eden uzmanlar
(odyologlar); konuşma patologları; ve / veya diğer sağlık
profesyonelleri.
Bu hastalarda genellikle rekonstrüktif cerrahi gereklidir.
Mandibular hipoplazisi olanlar, kaburga kemiği greftleri
kullanılarak rekonstrüksiyonlara sunulabilir ve az gelişmiş bir maksilla, kemik
distraksiyonu ve osteogenez ile uzatılabilir.
Dış kulağın rekonstrüktif ameliyatları 6 ila 8 yaşlarında
yapılabilir.
Daha hafif tutulumlu hastalarda ergenlik döneminde rekonstrüktif
çene ameliyatları yapılabilir.
Bu sendromlu çocuklar psikososyal zorluklar açısından daha
yüksek bir risk taşır, bu nedenle hastalara ve ailelere destek önerilir.
Prognoz, sistemik tutulumu olmayan komplike olmayan vakalarda
genellikle iyidir; Bununla birlikte, bazı ciddi vakalarda doğumdan
itibaren erken cerrahi müdahale gerekebilir.
Bu bölgelerdeki doğum kusurları riskindeki artış nedeniyle
böbrek ve kalp ultrasonları önerilebilir.
Hastalıkla İlişkili Genler
Aşağıdaki
bozuklukların belirtileri okülo-aurikülo-vertebral spektrumun belirtilerine
benzer olabilir. Karşılaştırmalar ayırıcı tanı için yararlı olabilir:
Treacher
Collins sendromu, kafatasının belirli kısımlarının (örneğin, supraorbital
jantlar ve elmopatik kemerler) ve alt çenenin az gelişmesine (hipoplazisi) bağlı
olarak kranyofasiyal bölgenin belirgin anormallikleri ile karakterize oldukça
nadir bir genetik hastalıktır. Treacher Collins sendromu ile ilişkili
semptomlar ve fiziksel özellikler vakadan duruma ciddi olarak değişebilse de,
kraniyofasiyal anormallikler elmacık kemiklerini, çeneleri, ağzı, kulakları ve
/ veya gözleri tutma eğilimindedir. Bu tür kranyofasiyal malformasyonlar
az gelişmiş (hipoplastik) veya eksik elmacık kemikleri; tamamiyle
gelişmiş, anormal derecede küçük bir alt çene (mandibular hipoplazi ve
mikrognati); alışılmadık derecede büyük bir ağız (makrostomi); ağız
çatısının malformasyonları (damak); ve / veya yanlış hizalanmış dişler
(maloklüzyon) gibi diş anormallikleri. Etkilenen bebekler ayrıca az
gelişmiş (hipoplastik) ve / veya kör biçimli veya kulak kepçeli dış kulak
kanallarına (atreziye) sahip olmayan (mikro) kulaklarda dışa dönük kulaklara
sahip olabilirler (işitme bozukluğu). Ek olarak, bozukluğu olan bebekler
aşağı doğru eğimli göz kapağı kıvrımlarına (palpebral fissüler), alt göz kapaklarının
dış üçte birinden kısmen veya tamamen doku yokluğuna (colobomas) ve / veya
ilave göz anormallikleri gösterebilir. Vakaların yaklaşık yüzde 40’ında,
Treacher Collins sendromu otozomal dominant kalıtıma sahiptir. Bununla
birlikte, vakaların yaklaşık yüzde 60’ında pozitif bir aile öyküsü
bulunamamıştır. Araştırmalar, bu gibi vakaların rastgele ortaya çıkan,
belirgin bir neden olmadan (sporadik) yeni genetik değişiklikleri
(mutasyonları) temsil ettiğini göstermektedir. (Bu hastalık hakkında daha
fazla bilgi için,
Erken
fetal gelişim sırasında birkaç kusurdan kaynaklanan nadir bir hastalık olan
CHARGE birlikteliği, vücudun birkaç organ sistemini etkileyen anormallikler ile
karakterizedir. CHARGE (C) gözün, özellikle gözün renkli kısmının (iris)
olobomasını temsil eden, iris’e anormal bir “anahtar deliği” şekli
veren bir kısaltmadır; (H) Fallot Tetralojisi, ventriküler ve / veya
atriyal septal defektler ve patent duktus arteriosus dahil olmak üzere toprak
defektleri; (A) burun arkasını boğaza bağlayan geçitlerin veya koana’nın
daraltılması veya tıkanması, yani normal burun nefesinin önlenmesi; (R)
bazı durumlarda zihinsel ve psikomotor geriliğin yanı sıra, büyüme ve
gelişmenin durması; (G) enital ve idrar anomalileri; ve (E) dış
kulakların ve orta kulakların kemiklerinin bozulmaması, östaki tüplerinin
yanlış çalışması, kulak kanallarının tıkanması ve / veya işitme kaybı gibi
anormallikler. CHARGE ilişkisinin tanısını doğrulamak için bu
karakteristik bulgulardan dördü mevcut olmalıdır. Klasik özelliklere ek
olarak, CHARGE Association ile bireyler ayrıca kranyofasiyal anormallikler,
böbrek ve merkezi sinir sistemi malformasyonları ve / veya trakeoözofageal
fistül ve / veya deliksiz anüs gibi diğer anormallikler de
gösterebilirler. CHARGE ilişkisinin kesin nedeni
bilinmemektedir; Bununla birlikte, çoğu vakanın rastlantısal olarak ortaya
çıktığı ve belirgin bir sebep olmadığı düşünülmektedir (sporadik).
Fetal
gelişim kusurlarından kaynaklanan nadir bir hastalık olan VACTERL birleşmesi,
vücudun birkaç organ sistemini etkileyen konjenital anormallikler ile
karakterizedir. VACTERL (V) hemivertebra ve alt vertebra (sakrum)
malformasyonu dahil olmak üzere ertebral anormallikleri temsil eden bir
kısaltmadır; (A) nal atrezi, anal açılmasının olmadığı bir durum; (C)
arter defektleri, özellikle ventriküler septal defektleri; (T) rakeo (E)
sifageal fistül; (R) böbrek ve hidronefroz yokluğu dahil enal
anormallikler; ve ön kol kemiklerinden birinin (radyal displazi) ve diğer
(L) imb defektlerinin yanlış gelişimi. Semptomlar çeşitli kombinasyonlarda
ortaya çıkabilir ve birçok bilinen bozukluğun tezahürü olabilir. Etkilenen
bireyler ayrıca vücudun diğer sistemlerini içeren ek anormallikler de
gösterebilirler. Çoğu durumda, VACTERL ilişkisinin, rastgele bir nedenden
ötürü rastgele meydana geldiği düşünülmektedir (sporadik); Bununla
birlikte, araştırmacılar bazı vakaların X’e bağlı veya otozomal resesif genetik
bir özellik olarak kalıtımsal olabileceğini öne sürmektedir.
Townes-Brocks
sendromu, doğumda görülen ve doğuştan görülen, nadir görülen kalıtsal bir
hastalıktır. Hastalıkla ilişkili semptomlar ve fiziksel özellikler vakadan
duruma geniş ölçüde değişebilse de, anormallikler yüz, kulaklar, kollar ve
bacaklar (uzuvlar), gastrointestinal sistem ve böbrekleri etkileme eğilimindedir. Bozukluğu
olan kişilerde, yüzün bir tarafı diğerinden daha küçük görünebilir (hemifasiyal
mikrozomi). Kulak anormallikleri, dış kulakların yanlış şekillenmesini,
aşırı cilt etiketlerini ve / veya kulakların önündeki girintileri (preauriküler
etiketler ve / veya çukurlar) ve / veya iç kulağın anormallikleri (duyusal
işitme kaybı) nedeniyle duyma bozukluğunu içerebilir. Etkilenen bireyler
ayrıca baş parmaklarında, ekstra parmaklarda (polidaktili), iki veya daha fazla
parmak ve / veya ayak parmağında (eş zamanlı olarak) bağlanma biçiminde
bozukluklara sahip olabilir. ve / veya diğer uzuv düzensizlikleri. Ek
olarak, Townes-Brocks sendromlu bireyler anal açıklığın yokluğunu gösterebilir
(deliksiz anüs); rektum ve cinsel organlar arasında anormal geçişler (rektovajinal
veya rektoperineal fistül); az gelişmiş böbrekler (böbrek
hipoplazisi); idrarın mesaneden geriye bir üretere (vezikoüreteral reflü)
aktığı bir durum; ve / veya diğer ilgili anormallikler. Ek olarak,
bazı durumlarda, etkilenen bireylerde kalp ve üreme organlarında anormallikler
de olabilir. Townes-Brocks sendromu otozomal dominant kalıtıma
sahiptir.
Branchio-oto-renal (BOR) sendromu, öncelikle kulakları, boynu
ve boğazı ve böbrekleri etkileyen anormalliklerle karakterize nadir görülen
kalıtsal bir hastalıktır. Etkilenen bireyler kulakların önünde aşırı cilt
etiketleri (preauriküler etiketler), orta ve iç kulağın malformasyonu, hatalı
biçimlendirilmiş dış kulaklar ve hafif ila şiddetli iletken ve / veya duyusal
işitme kaybı gösterebilir. Ek anormallikler, boğazdan boynun dış yüzeyine
(dal fistülü); bademcik bölgesinde anormal bir açıklık, kist veya
kitle; gözyaşı kanallarının (lakrimal kanalların) daralması (darlığı) ve /
veya yokluğu (aplazisi); ve / veya uzun, dar bir yüz, ağız çatısının eksik
kapanması (yarık damak), derin bir overbite ve / veya yüzün belirli kaslarının
felci dahil kraniofasiyal anormallikler. BOR sendromu olan kişilerde,
olağandışı şekilli böbrekler, böbreklerin toplama sisteminin çoğaltılması ve /
veya böbreklerin az gelişmişliği (hipoplazi) dahil olmak üzere, genellikle
hafif ila şiddetli böbrek (böbrek) anormallikleri
vardır. branşio-oto-renal sendrom otozomal dominant genetik özellik olarak
kalıtsaldır.
Nager sendromu,nadir görülen
kalıtımsal uyuşmazlığı karakterize eden bir hastalıktır ve kronofasiyal
malformasyon ile beraber başparmak, önkol anormalliklerini ortaya
çıkartmaktadır .Kronofasiyal malformasyon az gelişmişlikle oluşan; elmacık
kemikleri (yanak hipopalazisi) ile sonuçlanan aşağıya doğru meyilli olan
palpebral fissürler oluşturmaktadır. Eksik gelişim, aşağıya sıkışmış alt çeneye
(mandibular hipopalazi) neden olan anormal küçüklükte (mikroginati) ve küçük
mirkotia ve/veya kusurlu (displastik) çene, yarık dudak-yarık damak adı verilen
açıklıklara sebep olmaktadır (Mikroginati; solunum yolu problemlerine yol açar
ve tıkanıklığa sebep olur.) Dış kulaklarda (pinrus),genellikle kulak kanalı
tamamen tıkalı ya da kulak kanalı bulunmayan ve işitme bozukluğuna sebep olan
anomaliler ortaya çıkartır (iletken işitme kaybı).Nager sendromu diğer
sendromlardan; akrofasiyal disostoz, uzuv anomalileri, ayak başparmağı (radial
kısımda) yokluğu, az gelişmiş el ya da önkol (Radius kemiği) da görülen anormal
kemik füzyonları ile ayırt edilebilmektedir (radial sinostazis). Fakat el ve
ayak parmakları genelde normal durumdadır.Hastaların zekaları (mental)
etkilenmemiştir.Nager sendromu tipik olarak otozomal baskın gen ile (SB3F4
geni) değişiklere (mutasyonlara) neden olur.Nager sendromu birçok vakada rastgele meydana gelmesine
karşın; bireyin çocuklarına yeni bir gen değişimi olarak (de novo mutasyonu)
olarak iletilebilir.
Hastalığa Giriş ve Tanıtım
Nager sendromu tıp literatüründe ilk kez 1948’de Dr.Nager ve Dr.De Reynier tarafından tanımlanmıştır.Nager sendromu; akrofasiyal disostoz (AFDS) adında bilinen bir tür bozukluğa aittir.Bu bozukluklar kronofasiyal ve ekstremite anomalileri ile karakterize edilir.AFDS preaksiyal ve postaksiyal tip şeklinde iki çeşit bozulmayla ortaya çıkar.Nager sendromu preaksiyal durumda; kollarda,bacaklarda ve bu bölgelerin başparmakları üzerindeki kemiklerde (özellikle ayak başparmağı üzerinde) vücutta görülmektedir.
Hastalığın Belirti ve Bulguları
Bu tür spesifik semptomlar
aynı aileden olan insanlarda meydana gelmektedir (gen aktarımı).Bu durumdan
etkilenen bireylerde çeşitli kronofasiyal ve uzuv anomalileri (konjenital)
genellikle fark edilmektedir. Yaygın kronofasiyal anormallikler; az gelişmiş
elmacık kemiklerinde (malar hipopalazi),anormal derecede küçük bir alt çenede
(mikronati),ağzın üst iç damak kısmının yeterince kapatılamamasında (yarık
damak) veya velofarinjal yetmezlik (yumuşak damak,ağzın yeterince uygun şekilde
kapatılamaması,konuşulamaması)nda,burun boşluğunun arkasında daralma (koanal
atrezi),iç veya dış kulaklarda malformasyon,anomaliler dahilinde doku yokluğu
görülmektedir.Ek olarak kronofasiyal bulgular içerisinde; gözler aşağıya
sarkmaya eğimlidir (palpebral fissür), yani üst ve alt göz kapağı arasında
oluşan boşluk aşağıya doğru sarkmış durumdadır.Kolobom gözler (gözlerde çatlak
damar görünümü ve kızarıklık) alt göz kapaklarında kısmi veya tam olarak
kirpiklerin bulunmaması ,pitozis (sarkmış göz kapakları) ve bazı hastalarda
saçın elmacık kemiklerine, yanaklara kadar uzadığı görülmektedir.
Mikronati alt çene kemiğinin (mandibula)
az gelişmesine neden olur. Şiddetli mandibular hipopalazi,yarık damak ve koanal
atrezi ile birlikte bebeklik döneminde beslenme güçlüklerine yol açabilir.Bazı
durumlarda eğer komplikasyonlar tedavi edilmez ise hayatı tehdit eden solunum
sıkıntılarına sebebiyet verir.
Etkilenen/hasta olan bireylerde
temporomandibular eklem bozukluğu (TMJD) olabilir. Temporomandibular eklem
bozukluğunda; alt ve üst çene kafanın yanında birleşir ,çenede acıya neden
olur; yüz ve boyunda ağız kapatıldığında sert çene kaslarıyla birlikte üst ve
alt dişlerin doğru şekilde uyuşmadığı görülür (maloklüzyon).Etkilenen
bireylerde kulaklarda malformasyona (sakatlık durumu) neden olabilir ,bu
durumda genelde işitme kaybı ortaya çıkmaktadır .İletken işitme kaybı;iç, orta ve
dış kulakta dışardan gelen seslerin iletilmemesiyle oluşur.İşitme kaybı sorunu
derecelerle çeşitlendirilebilir .İşitme bozukluğu beraberinde konuşma
bozukluğunu da getirir ve geciktirir. Bireylerin Nager sendromuyla birlikte ekstra sahip
oldukları anomaliler de vardır ve bu anomaliler genellikle bireyin kollarını, ellerini,
ayak başparmaklarını etkilemektedir. Ayak başparmakları genellikle yoktur ya da
üçüncül fazladan bir ayak başparmağı gelişmektedir. Bahsedilen başparmağın
ekstra varlığında kemiğin kopyalanması (falanje) ve içinde (trifalanjel
başparmak); önkol kemiğinin üstünde yan tarafta bulunan (Radius) bölgeyi
etkilemektedir .Hastalarda daha az görülen anomalilerden biri de (sindaktili)
yapışık parmaklılık ve parmakların (kompodaktili) bükülmüş durumda
sıkışmalarıdır. Önkolun (radioulnar sinoztozis) anormal oluşumu iki ana kemikte
(ulna ve Radius) yumuşak doku bağlantısı şeklinde ayrıca oluşabilir. Bu
anomaliler önkolların anormal derecede kısa olmasıyla saptanabilir. Bu
anomalilere sahip bireyler günlük hayatlarında kollarını tamamen
düzleştiremedikleri için ve dirsek hareketleri kısıtlı olduğu için zorluk
çekmektedirler. Bu anomaliden şiddetli etkilenen bireylerin ise üst uzuvları
çok daha kısa durumdadır (fokomeli).Genellikle önkol ve ellerde yaygın
anomaliler olduğunu bilsek de ,bazı bireyler ayak ve bacaklarında benzer duruma
sahiptirler. Bu durumda daha önce de bahsedildiği gibi yapışık ayak parmakları ,ayak
başparmağının içe dönmesi (hallux valgus),ayak parmaklarının bulunmaması, clubfoot
görülmektedir.
Çoğu hasta Nager sendromuyla beraber
sağlıklı durumdadır.Fakat sendromdan şiddetli etkilenen bireylerde ciddi içsel
bozukluklar görülmektedir.Bu içsel bozukluklar özellikle böbrekleri ve/veya
kalb etkilemektedir.Tıp literatüründe ek olarak nadir semptomlar
raporlanmıştır.Buna örnek olarak;diafragmatik herni(diyafram fıtığı-göğüs ve
karın arasında anormallik),az gelişmiş larinks (gırtlak), sebebiyle solunum
problemleri,ek iskelet problemleri (ilk kaburganın az gelişmesi),omurganın
anormal derecede eğriliği (skolyoz) veya kalça çıkığı gösterilebilir.
Hastalığın Nedenleri
Çoğu durumlarda Nager sendromu
‘’SF3B4’’ geninde mutasyonlara sebep olur.Genler proteinleri oluşturmak için
talimat ve bu durum vücudun birçok fonksiyonunda kritik bir rol oynar.Mutasyon
gerçekleştiğinde genin protein ürünü hatalı,yetersiz ya da yok olmuş
olabilir.Bu fonksiyonlara bağlı olarak belirtili olan protein vücuttaki birçok
organ sistemini olumsuz etkileyebilir.Çünkü Nager sendromuna sahip bireyin ‘’SF3B4’’ geninin tek bir
kopyasında değişiklik vardır.Bu da bizim bildiğimiz gibi Nager sendromu bireye otozomal dominant gen koşulu ile
yerleşir.Çoğu durumda ise yeni bir sperm ve yumurta oluşumu sonunda ortaya
mutasyon çıkar.Bu mutasyon sadece aile fertleri arasına yeni katılacak olan
bireyin etkilenmesini;önceki ya da sonraki bireylerde etkilenme olmamasını
sağlar.Ancak ailede Nager sendromlu ilk
kişi olan birey yine de çocuklarına bu hastalığı aktarmada %50 riske
sahiptir.Görünüşe göre etkilenmeyen ebeveynler için doğmuş,Nager sendromlu kardeşlerin önceki raporları, Nager sendromu için farklı bir resesif formunu temsil
edebilir.Ancak orta derecede etkilenmiş bir ebeveyndeki durumu tanımadığı veya
sadece yumurtalık ve testiste gen değişikliği olan bir ebeveyn olması nedeniyle
daha olasıdır.(gonodal-germinal-mozaisizm)
Etkilenen Popülasyonlar
Nager sendromu kadınlarda ve erkelerde eşit olarak görülmektedir ve eşit olarak etkilemektedir.Genel popülasyonda hasalığın tam artışı ve yaygınlığı bilinmemektedir.Bu hastalık birçok vakasında yanlış teşhis edilmiş veya bir teşhis konulamamıştır;tıp literatüründe 100’den fazla vaka raporlanmış fakat genellikle hastalığın tanısının doğru frekansta belirlenmesi mümkün olamamıştır. Nager sendromu nadir bir hastalık olmasına rağmen akrofasiyal disostozdan en yaygın görülenidir.
Nager Sendromu ile İlgili Olan, Benzer Hastalıklar
Aşağıdaki yazılan
hastalıkların semptomları Nager sendromunun belirtilerine benzerlik göstermektedir.Fakat
ayrı bir tanı konulması için bileşimler yararlı olmaktadır.
Miller Sendromu; postaksiyal akrofasiyal disostoz olarak da bilinmektedir. Nadir bir genetik hastalıktır ve kronofasiyal malformasyonlar ile karakterize edilir .Kollarda, ellerde ve/veya ayaklarda; -genellikle postaksiyal- küçük parmak ve ayak parmağı tarafında anomaliler meydana gelmektedir .Kronofasiyal anomali; bireyde az gelişmiş elmacık kemikleri (malar hipopalazi),anormal derecede küçük bir alt çene (mikroginati),yarık dudak,küçük ve çıkıntılı kepçe kulalar ve/veya doku eksikliği, kolobomlara (düşük göz kapakları) sebebiyet vermektedir .Uzuvlarda anomalilere; eksik gelişme, ayak parmaklarında tek düzelik ve/veya yanlış gelişme ,kollarda anormal kemik füzyon oluşumu (radioulnar sinoztozis), kollarda alışılmadık derecede kısa görünüm oluşturmaktadır. Bazı durumlarda ek olarak fiziksel anomaliler ortaya çıkmaktadır fakat zeka bu durumdan etkilenmez. Miller sendromu Nager sendromuna bu semptomlarıyla çok benzese de asıl farklılık farklı genlerde mutasyonlar olması sonucuyla belirlenir. Miller sendromu; mutasyonların ‘’DHODH’’ geninde neden olduğu özellikler sayesinde otozomal resesif olarak miras alınır.(genler sayesinde aktarılır).
Treacher Collins Sendromu; nadir bir genetik hastalıktır ve kafa, yüz anomalileriyle Nager sendromundan (hipopalazik yüz yapıları, çene ve elmacık kemiği yakınındaki yapılar da dahil olmak üzere –elmacık kemiği kompleksi- ) sayesinde ayırt edilmektedir. Ayrıca bireyin uzuvları da normal durumdadır. Kronofasiyal anomalilerde ağızda, gözlerde veya çenede; elmacık kemiklerine dahil olma eğilimi bulunmaktadır.Ek olarak çeşitli yüz anomalileri; hastalıktan etkilenen bireyde kusurlu dış kulaklar ve orta kulak, göz (oküler) anomalileri; aşağıya doğru sarkmaya meyilli göz kapakları ve palpebral fissür meydana gelmektedir. Ayrıca hastalıktan etkilenen bireylerde işitme kaybı ve nefes alma (solunum) zorlukları gelişmektedir. Zeka ve davranışsal anomalilerde; raporlanmış bir durumun parçası olarak mikrosefali ve psikomotor gecikmeleri (delay) görülmektedir. ‘’TCS’’ ile ilişkili spesifik semptomlar ve fiziksel özellikler bir kişiden diğerine çok büyük ölçüde olabilir. Hastalığın tanısı konulmadan ve semptomları hafifken diğer belirtiler çok ciddi gelişebilir; hayatı tehdit eden ciddi bir solunum komplikasyonları ortaya çıkabilir. TCS; ‘’TCOF1, POLR1C, POLR1D’’ genlerinde meydana gelen bir mutasyondan kaynaklanır. TCOF1 veya POLR1D durumunda kalıtım şekli otozomal dominantken,POLR1C durumunda kalıtım otozomal resesiftir.
Rodriguez Sendromu; Rodriguez sendromu, Rodriguez’in akrofasiyal disostozu olarak da bilinir ve son derece nadir görülen bir hastalıktır. Rodriguez ve Nager sendromlarında da uzuv ve kronofasiyal anomalileri olarak ciddi bir benzerlik bulunmaktadır. Bazı araştırmacılar Rodriguez Sendromuna ait vakaların, Nager sendromunun daha ciddi bir ifadesi olabileceğine inanmaktadır. Ek olarak fiziksel bulgular çoğunlukla Nager sendromu ile birlikte bulunur. Rodriguez sendromlu bireylerde; omuz ve pelvik eklem desteği az gelişmiş durumdadır; kalp, merkezi sinir sistemi ve ürogenital sistemlerinde anormallikler meydana gelmiştir. Rodriguez sendromlu bebekler genellikle ölü doğarlar ya da yenidoğan dönemlerinde şiddetli solunum yolu problemleri nedeniyle direkt ölmektedirler. Rodriguez sendromunun kalıtımsal olduğu düşünülüyordu fakat son zamanlarda bir çocukta Rodriguez sendromuyla birlikte, SB3F4 geninin bir kopyasının de novo mutasyonuna uğradığı görüldü ( Nager sendromunda olduğu gibi). Çeşitli nadir bozukluklar; Wayers akrofasiyal disostoz, Catania akrofasiyal disostoz ve Palagnia akrofasiyal disostoz postaksiyal ve akrofasiyal disostoz bulgularını içermektedir. Diğer bozukluklar çok fazla semptomlara sahip POLR1C durumunda r ya da Nager sendromunun fiziksel bulgularıyla örtüşmektedir. Bunlara örnek; Ophthalmo akromelik sendromu, Pallister Hall sendromu, Mandibulafasiyal disostoz tipi Guion-Almeida, Burn-Mckeownsendromu ve Oculo-Auriculo-Vertebral sendromu gösterilebilir.
Teşhis
Nager sendromunun tanısı uzun
süre düşünülmüş bir klinik evrime dayanır, detaylı hasta öyküsü ve
karakteristik fiziksel bulguların belirlenmesiyle açıklanabilir. Bu hastalıkla
ilişkili anormalliklerin çoğu doğumda
bulunur (konjenital). Moleküler genetik testleri Nager sendromunun tanısının doğru yapılmasını
sağlar. Moleküler genetik testleri SF3B4 genindeki mutasyonu tespit edebilir
ama bu uygulama sadece uzman laboratuvarlarda gerçekleşmektedir.
Klinik Test ve Çalışma
Özel röntgen çalışmaları
hastalığın varlığını onaylar ve/veya kesin olarak kronofasiyal anormallikler
tarafından belirlenir.Örneğin bu tür görüntüleme testleri; anormal derecede
küçük olan çeneyi (mikroginati) , gelişmemiş olması sebebiyle alt çene kemiğini
(mandibular hipopalazi) ve az gelişmiş elmacık kemiğini (malar hipopalazi)
göstermektedir.
Standart Uygulanan Terapiler ve Hastalığın Tedavisi
Nager sendromunun tedavisi her bireyde görülen belirgin spesifik semptomlara göre uygulanır.Tedavi uzman ekibin koordineli çalışmasını gerektirir.Çocuk doktorları, ağız cerrahisinde uzmanlar, plastik cerrahi uzmanları, pediatrik kulak-burun-boğaz uzmanları (pediatrik KBB), göz bozukluklarının teşhis ve tedavisinin uzmanları (göz doktorları) kulak bozukluklarının teşhis (KBB Hekimleri), işitme kaybının tedavisinde uzmanlar (odyologlar), psikologlar ve diğer sağlık hizmeti profesyonelleri ; hasta bireyin tedavisini sistematik ve kapsamlı bir şekilde planlamalıdır.Hastalıktan etkilenen bireyler kraniyofasiyal merkeze yönlendirilmekten yararlanabilirler. Özel tedavi olarak; boğazda küçük bir kesik ile açıklık oluşturduktan sonra, boğaza belirli bir boyutta kesiğe uygun tüp yerleştirilir v, buradaki amaç hastanın solunum yolu açıklığını sağlamaktır (trakeostomi). Bireyin doğru beslenmesini sürdürebilmek için de cerrahi girişim gerekli olabilir ; midede küçük bir açıklık oluşturularak beslenme tüpü içeriye doğru salınır. Çenedeki, gözlerdeki ve uzuvlardaki anomalileri düzeltebilmek için cerrahi girişim sağlanmalıdır. Yarık damak ve yarık dudak mevcutsa eğer bu durumda ameliyat ve konuşma terapisi yapılır. Kaburgalarda oluşan anomalilergibi iskelet malformasyonlarında , dirsek hareketinin ve kolların hareketinin kısıtlılığında, skolyozda cerrahi işlem uygulanmalıdır. Konjenital (doğumsal) kalp sorunlarında/hastalıklarında da ameliyat durumu gereklidir. Erken müdahale ile uygun fiziksel,mesleki ve konuşma terapisi servisleri çocuklarda tam potansiyele ulaşmayı sağlatıyor.Fiziksel terapiler, yürüyüşlerinde yardımcı olmak için gerekli olabilir.Konuşma terapisi ise; işitme kaybı sebebiyle konuşma gelişimi gecikmesi olan bireylerin konuşmalarını sağlar.İşitme kaybı kulaklara tüplerin takılmasını gerektirebilir veya bir işitme cihazı kullanılmasını gerektirebilir. Etkilenen bireylere genetik danışmanlık tavsiye edilmektedir, aileleri de bu öneriden yararlanmalıdır. Tüm aile içinse aslında en gerekli olan psikososyal bir destektir.
Bartter sendromu, böbrek fonksiyonlarında spesifik kusurların
olduğu nadir bir genetik hastalık grubu için kullanılan genel bir isimdir. Bu
kusurlar böbreğin tuzu tekrar emme kabiliyetini bozar ve vücuttaki çeşitli elektrolit
ve sıvı konsantrasyonlarında dengesizliklere neden olur. Etkilenen
elektrolitler birincil olarak potasyum, kalsiyum, magnezyum, sodyum ve klorür
gibi mineral tuzlarıdır.
Bebeklik döneminden başlayarak, etkilenen bireyler genellikle
beklenen oranda büyümez ve kilo alamazlar. İdrarlarında fazla miktarda tuz
(sodyum klorür) kaybederek susuz kalmaya, kabızlığa ve artan idrar üretimine
(poliüri) yol açar. Ek olarak, idrarla çok miktarda kalsiyum atılması,
kemiklerin zayıflamasına neden olur. Bartter sendromu
ayrıca kandaki düşük potasyum ile karakterizedir, bu da kas güçsüzlüğü, kramp
ve yorgunluk ile sonuçlanabilir. Nadiren de olsa, etkilenen çocuklarda, iç
kulak anormalliklerinin neden olduğu işitme kaybı gelişir.
Genetik Değişiklikler/ Etken
Faktörler ve İlgili Genler
Hastalığın mutasyona uğrayan genlere göre beş alt bölümü
vardır.
Bartter
sendromu tip 1 (SLC12A1 geni)
Bartter
sendromu tip 2 (KCNJ1 geni)
Bartter
sendromu tip 3 (CLCNKB geni)
Bartter
sendromu tip 4A (BSND geni)
Bartter
sendromu tip 4B (CLCNKA ve CLCNKB genleri)
Bartter
sendromu tip 5 (MAGED2 geni)
Bartter sendromu ile
ilişkili genler normal böbrek
fonksiyonunda önemli roller oynar. Bu genlerden üretilen
proteinler böbreklerdeki tuzun yeniden emiliminde rol oynar. Bu genlerin
herhangi birindeki mutasyonlar böbreklerin tuzu yeniden emme yeteneğini bozar
ve idrarda aşırı tuz kaybına neden olur.
Belirti ve Semptomlar
Bartter sendromu için başlangıç yaşı, hastalık şiddeti ve
gözlemlenen spesifik semptomlar aynı alt tipte olan kişiler arasında bile
kişiden kişiye büyük ölçüde değişebilir. Bazı kişilerde hafif semptomlar
gözlemleneblirken; diğerleri doğumda ciddi, potansiyel olarak yaşamı tehdit
edici komplikasyonlar yaşayabilir.
Sık görülen semptomlar; kas güçsüzlüğü, kramp, spazm ve
yorgunluktur.Bazı vakalarda aşırı susuzluk (polidipsi), aşırı idrara çıkma
(poliüri) ve gece idrara çıkma ihtiyacı (nokturia) da görülebilir. Aşırı sıvı
alımı yüzünden, sık idrara çıkma dehidratasyona(sıvı kaybı) neden olabilir.
Meydana gelebilecek ek semptomlar kabızlık, kusma, yüksek vücut ısısı,
uyuşukluk ve genel sağlıksızlık hissini içerir. Çocuklar büyüdükçe, büyüme
hızları yaşlarına ve cinsiyetlerine bağlı olarak beklenenin altında (büyüme
geriliği) olabilir. Eğer tedavi edilmezse, etkilenen bireyler yetişkinlerden
beklenenden daha kısa olabilir (kısa boy). Bazı çocuklar gelişimsel dönüm
noktalarına ulaşmada gecikmeler yaşayabilir. Etkilenen bazı bebekler, üçgen
şeklinde bir yüz, belirgin alin, büyük gözler, belirgin, sivri kulaklar ve
ağzın köşelerinin sarkması nedeniyle somurtma ifadesi gibi karakteristik yüz
özelliklerine sahip olabilir. Önemli elektrolit dengesizlikleri yaşayan bazı
kişilerde, düzensiz kalp atışları (kalp ritmi) veya kas zayıflığı (felç) gibi
ciddi komplikasyonlar gelişebilir. Nadir olmasına rağmen, tedavi edilmezse, bu
kardiyak aritmiler, ani kalp durması ve potansiyel olarak ani ölüme neden olmak
için potansiyel olarak ilerleyebilir.
Genel olarak, Bartter sendromu tip 1, 2, 4a ve 4b, erken
başlangıç yaşı ve daha ciddi semptomlar ile ilişkilidir. Bartter sendromu tip 1
ve 2 olan hastalar tipik olarak idrarda yüksek seviyede kalsiyum içerir ve
böbreklerde kalsiyum birikmesine neden olabilir. Böbreklerdeki kalsiyum
birikimi (kalsifikasyon) sonunda böbrek fonksiyonunu etkilenebilir. Bartter
sendromu tip 4A ve 4B’de, işitsel sinirlerin beyne duyusal girdi iletebilme
yetersizliği nedeniyle etkilenen bebeklerde doğuştan sensorinöral sağırlık
görülür.
Bartter sendromu tip 3 doğumdan önce de ortaya çıkabilir,
ancak genellikle daha hafif semptomlarla ilişkilidir ve bu alt tipte birçok
hastada bebeklik döneminde veya erken çocukluk döneminde büyüme problemleri ile
görülür.
Bartter sendromu tip 1, tip 2 ve Bartter sendromu tip 4a ve
4b alt gruplarında belirti ve semptomları doğum öncesi dönemde görülebilir. Uterodaki
anormal böbrek fonksiyonu aşırı idrar üretimine ve gelişmekte olan fetüsün
etrafında anormal bir amniyotik sıvı birikmesine neden olabilir.
(polihidroamniyoz) Genellikle erken doğum görülür. Yenidoğan döneminde,
etkilenen bebeklerde aşırı idrara çıkma (poliüri) ve hayatı tehdit eden ateş ve
dehidrasyon olayları görülebilir.Kusma ve ishal de görülebilir.
Özel bir form Bartter tip 5; tipik olarak erken doğuma yol
açan şiddetli amniyotik sıvı (polyhydramnios) ile kendini gösterir. Bazı
durumlarda, prematürite o kadar erkendir ki bebek anne rahminin dışında
yaşamayabilir. Ancak, doğduktan sonraki haftalarda, yüksek idrar çıkışı ve
elektrolit kaybının tüm böbrek semptomları, tedaviye ihtiyaç duymadan
kendiliğinden düzelir. Bu durumun altında yatan gen X-kromozomundadır, bu
nedenle Bartter sendromu tip 5 baskın olarak erkeklerde görülür.
Genetik Görülme Sıklığı
Bartter sendromları erkekleri ve kadınları eşit sayıda
etkiler.Genel nüfusta yaklaşık 100.000 kişiden birini etkileyeceği tahmin
edilmektedir. Bununla birlikte, birçok vaka teşhis edilmez veya yanlış teşhis
edilir, bu da genel popülasyondaki gerçek sıklığı belirlemeyi zorlaştırır.
Bartter sendromları herhangi bir ırk veya etnik kökene sahip bireylerde ortaya
çıkabilir.
Kalıtım Paterni/Deseni
Hastalık tip 5 hariç bütün alt tiplerinde otozomal resesifpaternde
kalıtsaldır.Bu, her hücrede genin her iki kopyasında da mutasyon olduğu
anlamına gelir. Ebeveynlerin ikisi de hastalığı taşır ama izlerini
göstermezler. Tip 5 alt türü ise X’e bağlı resesif kalıtılır.
Teşhis Yöntemleri ve
Tedaviler
Bartter sendromlarından birinin tanısı, karakteristik
semptomların tanımlanması, ayrıntılı bir hasta geçmişi, ayrıntılı bir klinik
değerlendirme ve çeşitli uzmanlık testlerine dayanır.
Bu bozuklukları teşhis etmek için kullanılan laboratuvar
testleri, serum elektrolit seviyelerini, özellikle potasyum, klorür,
bikarbonat, magnezyum, renin ve aldosteron seviyelerini belirlemek için kan
testlerini ve sodyum, klorür, potasyum, kalsiyum ve magnezyum da dahil olmak
üzere idrar elektrolitlerinin ve prostaglandin E2 varlığını belirlemek için
idrar testleri.içerir. Son olarak moleküler genetik testlerle de, Bartter
sendromlarına neden olduğu bilinen belirli genlerdeki mutasyonlar tespit
edilebilir ve tanı doğrulanabilir.
Bartter sendromlarının tedavisi, her bir bireyde belirgin
olan spesifik semptomlara yöneliktir.Tedavi, uzman bir ekibin koordine
çabalarını gerektirebilir. Tedavi belirli takviye ve ilaçların ömür boyu
uygulanmasını gerektirir. Tedavinin dayanak noktası vücuttaki sıvı ve
elektrolitlerin uygun dengesini restore etmektir. Bu tipik olarak elektrolit
dengesizliklerinin düzeltilmesine yardımcı olmak için sodyum ve potasyum klorür
desteğini içerir. Tedavinin yanında büyüme hormonu takviyesi bazı vakalarda
büyüme geriliği ve Bartter sendromuyla ilişkili kısa boylanma tedavisi için
uygulanabilir.Ayrıca özellikle Bartter sendromu tip 3’te, magnezyum takviyesi
kas spazmlarını tedavi etmek için kullanılabilir.
Etkilenen bireylerin yeterli tuz ve su alımı gereklidir.
Hailey-Hailey Hastalığı (İyi Huylu
Kronik Pemfigüs)
Genel Bilgi
Hailey-Hailey
hastalığı nadir ve genetik bir cilt hastalığıdır. Pemfigüs, ciddi bir otoimmün
hastalığıdır ve ağrıya sebep olan deride su kabarcıklarına neden olur.
Semptomlar yaz ayında sıcaklıktan ve terden dolayı daha da kötüleşebilir.
Deride kırmızı kabarcık ve kaşıntı en çok koltukaltında, boyunda, göğüste ve
kasıkta görülmektedir. İltihaplanıp şişmiş bölgeler kaşınabilir ve yanabilir. Hastalıktan
etkilenen bireylerin güneş yanığına karşı kendilerini korumaları önerilir. Çok
nadir olarak derideki bu yaralar skuamoz hücreli karsinom cilt kanserine sebep
olabilirler.
Genetik Değişiklikler/Etken Faktörler
Hailey-Hailey
hastalığına ATP2C1 genindeki mutasyonlar sebep olmaktadır. Bu gen, kalsiyum ve
magnezyum pompası olan proteinin oluşumu için önemli rol oynar. Eğer kalsiyum
pompasının fonksiyonu bozulursa hücreler birbirine bağlanamz ve deriye zarar
verir. Bu protein en çok keratinositlerde (derinin dış katmanı olan epidermisteki
ana hücreler) aktiftir. Keratinnositlerin birbirine bağlanmasında problem
olursa, bu deride su kabarcığına sebep olur.
Belirti ve Semptomlar
Deride
erozyon (deride sınırlı bir bölgede epitel kaybı), deride kabarcıklar, deride
kaşıntı, deride iltihaptan dolayı kızarıklık, deri yüzeyinde aşırı keratin
oluşması ve derinin kalınlaşması, epidermiste bulunan hücreler arasında
bağlantının kopması. Derideki yaralar sarı bir yığılım oluşturabilir.
Yaralardaki enfeksiyonlar kokuya sebep olabilir.
Genetik Görülme Sıklığı
Çok
nadir olduğu için yaygınlığı bilinmemektedir ama tahminlere göre 50 bin insanda
1 kişiyi etkilemektedir. Erkek ve kadınları eşit oranda etkilemektedir.
Kalıtım Paterni/Deseni
Otozomal dominant geçişli bir
hastalıktır.
Teşhis Yöntemleri ve Tedaviler
Teşhis
için deri dokusuna biyopsi yapılabilir. Genetik test ile ATP2C1 genindeki
mutasyonlar belirlenebilir. Bu hastalık için bir tedavi yoktur, sadece semptomlara
ayrı ayrı özel olarak tedavi uygulanıp semptomların azalmasına yardım
edilebillir. Güneş koruyucu ve nemlendirici kremler kullanılması önerilir.
Deride enfeksiyon gelişirse antibiyotik tedavisi uygulanır. Kortikosteroid
kremler semptompların hafiflemesine yardımcı olabilir. Botulinum toxin, karbondioksit lazer ile ablasyon
(CO2 ablation laser) ve dermabrazyon (dermabrasion) üstünde
durulan tedavilerdir ve etkili sonuçlar alınmıştır.
Hastalıkla İlişkili Genler
ATP2C1 genindeki mutasyonlar bu hastalığa sebep
olmaktadır.
Mukopolisakkaridoz
tip I (MPS I), vücudun birçok bölümünü (multisistem) etkileyen nadir bir
genetik hastalıktır. MPS I, en erken yaşta ortaya çıkan şiddetli formlardan
çocuklukta çok belirgin hale gelmeyen daha az şiddetli formlara kadar değişen
bir hastalık yelpazesi olarak düşünülür. Hurler sendromu hastalığın erken
yaşlarda ortaya çıkan en şiddetli formudur. Hurler sendromu; iskelet anormallikleri,
bilişsel bozukluklar, kalp rahatsızlıkları, solunum problemleri, genişlemiş
karaciğer ve dalak, karekteristik surat ile karakterize edilmiştir.
Genetik Değişiklikler ve Etken Faktörler
Hurler sendromunda IDUA
genindeki (4p16.3) mutasyon glikozaminoglikanların (GAGs) yıkılmasında rol
oynayan alfa-L-iduronidaz enziminde eksikliğe yol açarak dermetan sülfat ve
heperan sülfatın lizozomal birikimine yol açar.
Belirti ve Semptomlar
Şiddetli MPS I yani
Hurler sendromu, çoklu organ ve dokuları içeren kronik ve progresif bir
hastalık seyri ile karakterizedir .
Hurler sendromlu çocuklar doğumda normal görünür Ancak kasık veya göbek fıtıkları olabilir. Karakteristik görünüm yaşamın ilk yıllarında gelişir.
Şiddetli MPS I için ortalama tanı yaşı yaklaşık dokuz aydır.
Yüz özelliklerinin 3-6
aylıkken hafifçe kabalaşması genellikle tespit edilen ilk anormalliktir.
Kafa büyük ön kemikler yüzünden şişkindir.
Kalp kapağı
anormallikleri, beyinde sıvı birikmesi, genişlemiş bir karaciğer ve dalak (hepatosplenomegali)
ve geniş bir dil (makroglossia) görülür. Bazı vakalarda sık sık üst solunum
yolları enfeksiyonları ve uyku apnesi
görülebilir.
Progresif
hepatosplenomegalinin neden olduğu karın şişkinliği yaygındır. Organ
büyümesi görülse de, karaciğerde ve dalakta glikozaminoglikanların depolanması
organ işlev bozukluğuna yol açmaz.
Kornea bulanıklığı
yaygındır. Hastalık önemli görme kaybına neden olabilir. Etkilenen
kişilerde işitme kaybı ve tekrarlayan kulak enfeksiyonları da olabilir.
Figür: Mukopolisakkaridoz tip I ile ilişkili kornea bulanıklığı
Üç yaşına gelince
doğrusal büyüme azalır. Omurga gövdelerinin kusurlu ossifikasyon
merkezleri, düzleşmiş ve gagalanmış omurlara ve sonrasında da omurga
deformitesine yol açar. Bu bazı vakalarda hareketliliği engeller. Çoklu iskelet anormallikleri anlamına gelen distostoz multipleksler görülür.
Figür: Mukopolisakkaridoz tip I ile ilişkili kavisli omurga
Karpal tünel sendromu
bu bozukluğu olan birçok çocukta
gelişir ve el ve parmaklarda uyuşma, karıncalanma ve güçsüzlük ile karakterize
edilir.
Erken psikomotor gelişim
normal olsa da, gelişimsel gecikme 18 aylıkken genellikle belirgindir. Entelektüel
kapasitede ölçülebilir bir azalma bundan sonra aylık olarak gerçekleşir. İlerleyen dönemlerde zihinsel yeteneklerde
yavaş bir düşüş gerçekleşir. Sekiz ila on yaşları arasındaki ölüm zamanlarında,
çoğu çocuk zihinsel olarak engellidir.
Genetik Görülme Sıklığı
MPS1’in Hurler alt
türünün yaygınlığının Avrupa’da 1 / 200.000 olduğu tahmin edilmektedir.
Hastalıkla İlişkili Genler
Hurler sendromu; 4p16 kromozomu üzerindeki alfa-L-iduronidaz kodlayan
IDUA geninde homozigoz veya bileşik heterozigoz mutasyondan kaynaklanır.
Kalıtım Paterni
Hurler sendromu otozomal resesif kalıtılan genetik bir hastalıktır. Otozomal
resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış
genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve
semptomlarını göstermezler.
Klinik bulgular hastalık
şiddetine göre değişir. Ancak sadece klinik bulgular hastalık tanısı için
yeterli değildir. Bu yüzden laboratuvar analizleri yapılır.
Laboratuvar analizleri, idrarda
artan glikozaminoglikanlarının yani heparan ve dermatan sülfatın kalitatif (GAG
elektroforezi) ve kantitatif (toplam idrar ürronik asit ölçümü) analizine
dayanır. Ancak Ne nicel ne de nitel yöntem, MPS I de dahil olmak üzere spesifik
bir lizozomal enzim eksikliğini teşhis edemez; ancak, yöntemlerden biri veya
her ikisi ile tespit edilen bir anormallik, bir MPS bozukluğunun muhtemel
varlığını gösterir.
Kesin tanı için
moleküler genetik testler yapılabilir. Moleküler test yaklaşımları; IDUA
geninde Gen hedefli silme/ çoğaltma yapılmasına ya da Dizi analizine dayanır.
Son bir teşhis yöntemi
olarak da Alfa-L-iduronidaz enzim
aktivitesi çoğu dokuda ölçülebilir; tipik olarak periferik kan
lökositleri, plazma veya kültürlenmiş fibroblastlar kullanılır. Neredeyse tüm
MPS’li bireylerde, teşhis için kullanılan standart numunelerde tespit
edilebilir bir enzime rastlanaz.
Mukopolisakkaridoz tip I
tanısı, karakteristik semptomların tanımlanması, ayrıntılı bir hasta ve aile
öyküsü, ayrıntılı bir klinik değerlendirme ve çeşitli uzmanlık testlerine
dayanır. Karakteristik erken belirtileri olan bebeklerde tanıdan
şüphelenilebilir. Halen MPS I için yenidoğan taraması birçok alanda uygulanmaktadır.
Tedavi
1. Eksik enzimin değiştirilmesi: Lizozomal
enzimler, hücreler tarafından alınabildikleri ve kullanılabildiklerinden dolayı
eşsiz proteinlerdir. Bu nedenle, MPS I’yi tedavi etmenin potansiyel bir
yolu, hastalara eksik aldıkları alfa-L-iduronidaz enzimini vermektir. Bu 2
şekilde yapılabilir.
a. Bunlardan biri, enzim replasman tedavisi (ERT) olarak da
bilinen saflaştırılmış enzim ile beslemektir. Enzim replasman tedavisi, eksik
enzimin alfa-L-iduronidazın genetik olarak tasarlanmış (rekombinant) bir formla
değiştirilmesini içerir. Tüm Hurler hastalarına ERT önerilmektedir ve nörolojik
olmayan semptomları hafifleten yaşam boyu bir terapidir. ERT’nin erken
kullanımının, bu durumun bazı klinik özelliklerinin gelişimini geciktirdiği
veya hatta engellediği gösterilmiştir. Bu, enzimi haftada bir intravenöz
yoluyla bir hastaya infüze etmek suretiyle gerçekleştirilir.
b. Hematopoetik kök hücre nakli (HSCT), hayatta kalma süresini
uzatabildiği, nörobilişimi koruyabildiği ve bazı somatik özellikleri
iyileştirebileceği için, 2.5 yaşın altındaki (ve bu yaş sınırının üzerindeki
seçilmiş hastalarda) Hurler sendromlu hastalar için tercih edilen tedavi
yöntemidir.HSCT, hastalığın seyrinde, gelişimsel bozulma başlamadan önce erken
yapılmalıdır. HSCT sağkalımı artırabilir, büyümeyi artırabilir, yüz kalınlığını
ve hepatosplenomegaliyi azaltabilir, işitmeyi iyileştirebilir ve kalp ve
solunum semptomatolojisinin doğal geçmişini değiştirebilir. HSCT’nin iskelet ve
eklem belirtileri veya kornea bulanıklığı üzerinde daha az etkisi vardır. HSCT,
transplantasyon sırasında hafif fakat anlamlı olmayan bilişsel bozulma olan
çocuklarda bilişsel gerileme sürecini yavaşlatabilir. HSCT ile ilişkili
morbidite ve mortalite nedeniyle, günümüzde öncelikle ağır MPS I olan çocuklar
için önerilmektedir.
2. Hastalığın
spesifik semptomlarını hafifletmek: Mukopolisakkaridoz tip I tedavisinin önemli
bir bileşeni, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Bu,
uzmanlardan oluşan bir ekibin koordine çabalarını gerektirir
3. Etkilenen
bireyler ve aileleri için genetik danışmanlık önerilir. Tüm aile için
psikososyal destek de şarttır.
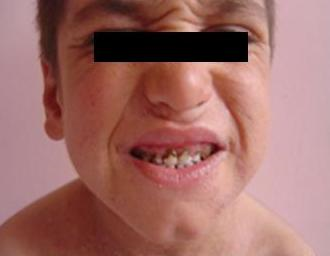
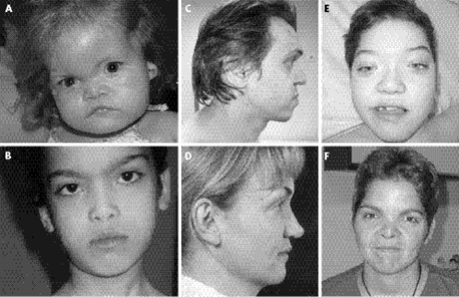
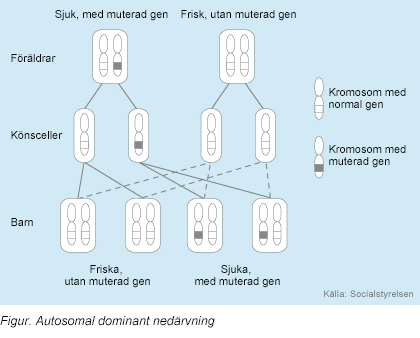
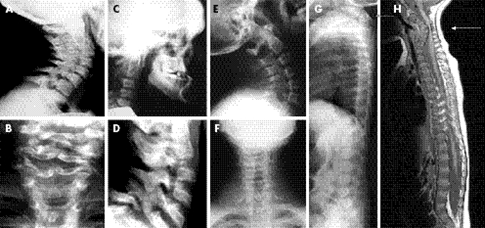
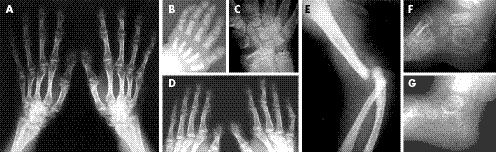
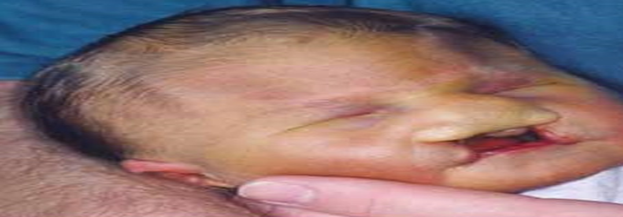
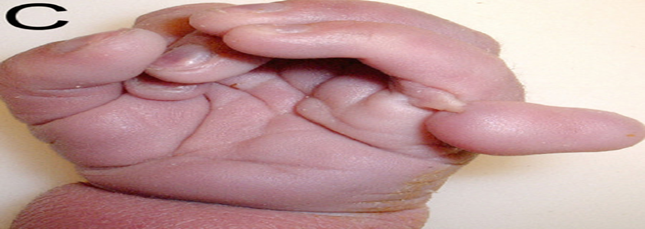
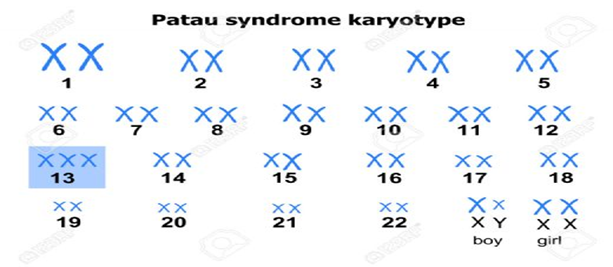
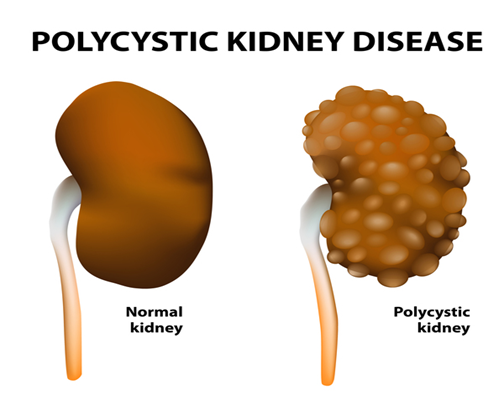
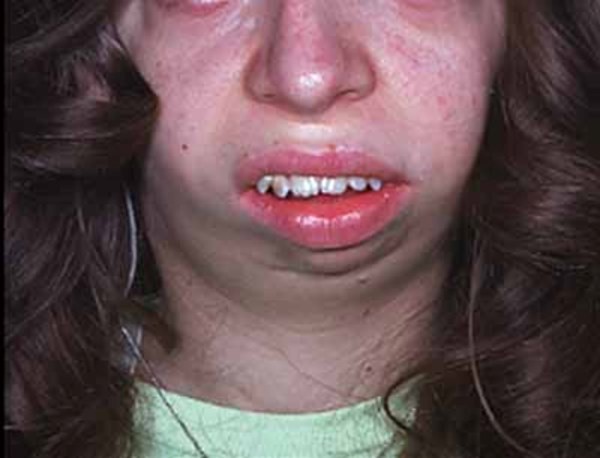
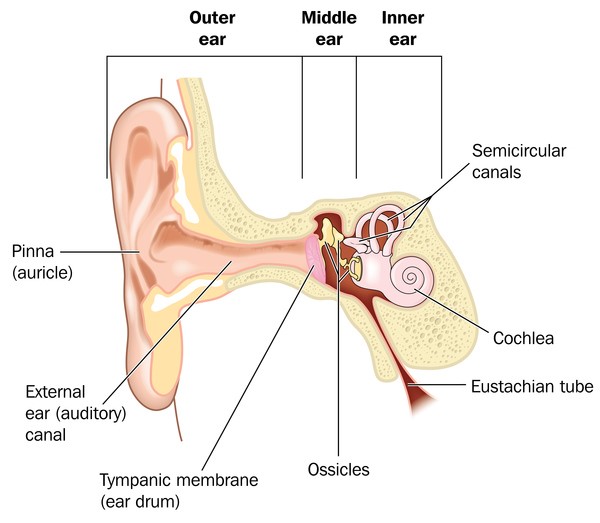
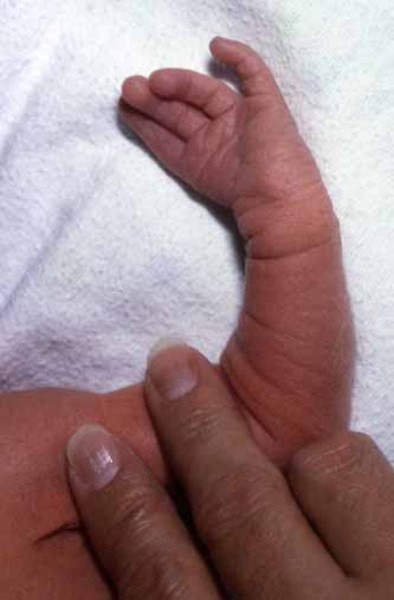
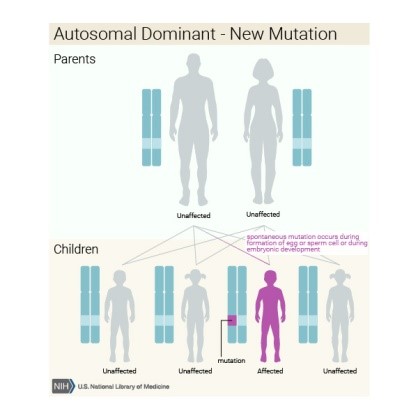
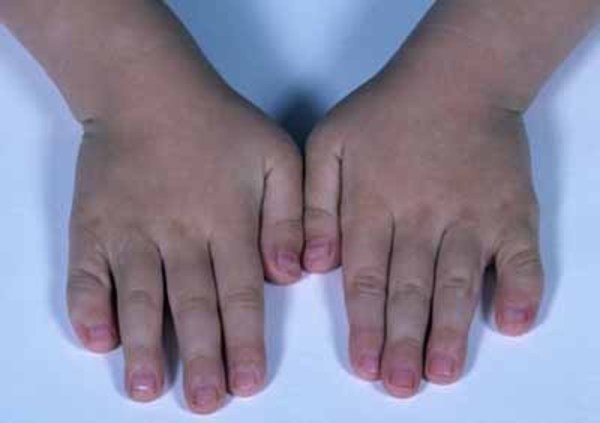
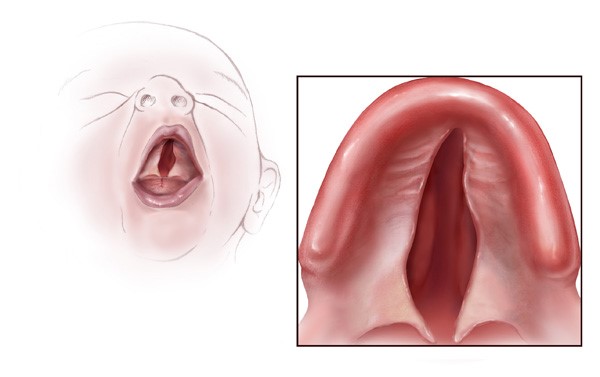
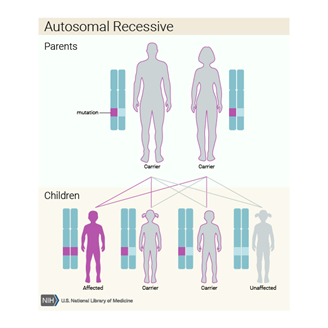
{kind=link}