Schinzel Giedion Sendromu (SGS); doğumla görülen, oldukça
nadir ve ciddi semptomları olan sistemik bir rahatsızlıktır. Bu sendroma sahip
çocuklar genellikle doğduktan sonra ancak birkaç yıl yaşayabilmektedirler.
Genetik
Değişikler/ Etken Faktörler
Sendrom, SETBP1 geninde meydana gelen bir mutasyon
sonucu görülmektedir. Bu genin, vücutta SET Bağlayıcı Protein 1’i kodladığı
bilinse de hakkında daha fazla bilgi halen aydınlatılmayı beklemektedir.
Belirti
ve Semptomlar
Öne doğru çıkık alın, yüzün ortasında düzlük ve kısa ve ucu yukarı bakan
bir burun ile karakterize olan özel bir yüz şekli
Hidronefroz
Ciddi gelişme ve zeka geriliği.
Kısa ekstremiteler, içbükey ya da dışbükey ayak anomalisi, kafatası ve
kaburga anomalileri gibi çeşitli iskelet sistemi deformiteler
İnmemiş testis, normalden küçük penis, normalden küçük uterus, az gelişmiş
iç ve dış vulva katlantıları, yanlış yerleşimli anüs gibi genitoüriner
anomaliler
Kalpte septal defektler, kapak anomalileri, az gelişmiş karıncıklar ya da
Patent Ductus Arteriosus
Nöbetler
Görme ve duymada çeşitli problemler
Tümör geliştirmeye yatkınlık
Kıllanma artışı
Tırnak anomalileri
Genetik
Görülme Sıklığı
Oldukça nadir olan sendromun, prevalansı belirsizdir.
Kayıtlı yaklaşık 50 vaka vardır.
Kalıtım
Paterni
Schinzel-Giedion Sendromu, SETBP1 genindeki yeni mutasyonlar
sebebiyle ortaya çıkmaktadır. Hastaların öykülerinde hastalığa sahip aile
bireyi bulunmamaktadır.
Teşhis
Yöntemleri/ Tedaviler
Teşhis yöntemi olarak genetik testler kullanılabilir. Ayrıca
prenatal ultrasonda hidronefroz görülmesi, bu sendromu olası bir tanı olarak
akla getirmelidir. Sendromu tedavi etmek mümkün değildir ancak semptomlara
yönelik tedaviler ve palyatif bakım hastanın yaşam kalitesini arttırabilir.
Krabbe hastalığı (ayrıca globoid
hücre lökodistrofisi olarak da bilinir) nadir görülen bir genetik hastalık
olup, ciddi bir nörolojik durumdur. Miyelin kaybından
kaynaklanan lökodistrofiler olarak bilinen bir grup bozukluğun parçasıdır.sinir
sisteminde (demiyelinizasyon). Miyelin, sinir sinyallerinin hızlı
iletimini sağlayan sinir hücrelerinin etrafındaki koruyucu kaplamadır. Krabbe hastalığı ayrıca beyinde, genellikle
birden fazla çekirdeğe sahip büyük hücreler olan
globoid hücreler adı verilen anormal hücrelerle de karakterize edilir..
İnfantilite adı
verilen Krabbe hastalığının en
yaygın şekli genellikle 1 yaşından önce başlar. İlk belirtiler ve
semptomlar tipik olarak sinirlilik, kas zayıflığı, beslenme güçlüğü, herhangi
bir enfeksiyon belirtisi olmadan ateş dönemleri, sert duruş ve gecikmiş
zihinsel ve fiziksel gelişimdir. Hastalık ilerledikçe, kaslar zayıflar ve
bebeğin hareket etme, çiğneme, yutma ve nefes alma kabiliyetini
etkiler. Etkilenen bebekler ayrıca görme kaybı ve nöbet
geçirir. Durumun ciddiyeti nedeniyle, doğar doğmaz belirtileri başlayıp teşhis
konmuş olan çocuklar 2 yaşına gelmeden ölür. Daha az yaygın olarak, Krabbe hastalığı çocukluk, ergenlik veya
yetişkinlikte başlar (geç başlangıçlı formlar). Görme problemleri ve
yürüme güçlüğü, hastalığın bu formlarında en sık görülen başlangıç
semptomlarıdır, ancak, semptomlar ve semptomlar etkilenen bireyler arasında
önemli ölçüde farklılık gösterir. Geç başlayan Krabbe hastalığı olan kişiler, durum başladıktan
yıllar sonra hayatta kalabilir.
Genetik Değişiklikler/ Etken Faktörler
GALC genindeki mutasyonlar Krabbe
hastalığına neden olur . Bu gen, galaktolipidler adı
verilen bazı yağları parçalayan galaktosilseramidaz adı verilen bir enzim yapmak
için talimatlar sağlar. Galaktosilseramid denilen galaktosilseramidaz
tarafından parçalanan bir galaktolipid, miyelinin önemli bir bileşenidir.. Galaktosilseramid’in parçalanması,
miyelinin yaşam boyunca meydana gelen normal cirosunun bir
parçasıdır. Miyelinin üretimi sırasında oluşan psikosin adı verilen bir
başka galaktolipid, galaktosilseramidaz tarafından parçalanmadığı takdirde
toksiktir.
GALC gen mutasyonları
galaktosilseramidaz enziminin aktivitesini ciddi şekilde azaltır. Sonuç
olarak, galaktosilseramid ve psikosin parçalanamaz. Aşırı
galaktosilseramid, belirli hücrelerde birikir ve globoid hücreler
oluşturur. Bu galaktolipidlerin birikmesi, miyelin oluşumunu bozan ve
sinir sisteminde demiyelinizasyona yol açan miyelin oluşturan hücrelere zarar
verir. Miyelin olmadan, beyindeki sinirler ve vücudun diğer bölümleri
sinyalleri düzgün şekilde iletemez ve bu da Krabbe
hastalığının belirtileri ve semptomlarına yol açar .
Belirti ve Semptomlar
Krabbe hastalığının belirti ve semptomları farklı yaşlarda gelişebilir. Erken başlangıçlı (infantil) Krabbe hastalığından etkilenen bebekler tipik olarak yaşamın ilk altı ayında özellikler geliştirirken, geç başlangıçlı formlardan etkilenen insanlar çocuklukta, erken ergenlikte ve hatta yetişkinlikte geç saatlere kadar semptom geliştirmeyebilir.
Krabbe hastalığından
etkilenen kişilerin yaklaşık% 85-90’ı, aşağıdaki özelliklerle karakterize edilen
infantil formdadır:
Daha sonra başlayan
formların belirti ve semptomları oldukça değişkendir ancak kas güçsüzlüğü ve
sertliği içerebilir; yürüme güçlüğü; görme kaybı; entelektüel
gerileme; ve / veya nöbetler.
Genetik Görülme Sıklığı
Amerika Birleşik
Devletleri’nde Krabbe
hastalığı 100.000 kişiden 1’ini etkiler. İsrail’de izole edilmiş birkaç toplulukta
daha yüksek bir insidans (1.000 kişi başına 6 vaka) bildirilmiştir. İskandinav
kökenli insanlar arasında en yaygın olanıdır.
Kalıtım Paterni/Deseni
Bu durum otozomal
resesif paternde kalıtsaldır, her hücrede genin her iki kopyasında da
mutasyon olduğu anlamına gelir. Otozomal resesif hastalığı olan bir
bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır,
ancak bunlar (taşıyıcı) genellikle durumun belirtilerini ve semptomlarını
göstermezler.
Teşhis Yöntemleri ve Tedavileri
Krabbe hastalığı,
vücuttaki çeşitli hücrelerde ve dokularda zararlı miktarlarda lipitlerin biriktiği ve beyin hücrelerini tahrip ettiği
nadir, kalıtsal bir metabolik hastalıktır.
Anormal bir yenidoğan taramasından dolayı karakteristik belirti
ve semptomların varlığına bağlı
olarak Krabbe hastalığının tanısından
şüphelenilebilir . Ardından teşhisi onaylamak için ek testler
istenebilir. Bu test genellikle galaktosilseramidaz seviyelerini
değerlendirmek için bir kan testi ve / veya cilt
biyopsisi içerir.
Ne yazık ki, Krabbe
hastalığının tedavisi yoktur. İnfantil Krabbe hastalığı genellikle iki yaşından
önce ölümcüldür. Bununla birlikte, ön çalışmalar hematopoetik olduğunu
göstermektedir; kök hücre nakli (yani göbek kordon kanı hücreler)
henüz belirtileri gelişmemiş etkilenen bebeklerde ve hafif belirtileri olan
yaşlılarda etkili bir tedavi olabilir. Örneğin, bu tedavinin hastalığın
ilerlemesini geciktirebileceğine ve sağkalım ve yaşam kalitesini
artırabileceğine dair kanıtlar vardır. Hem kısa hem de uzun vadeli
faydalar bildirilmiş olmasına rağmen, veriler öncelikle küçükklinik denemeler
düzeyindedir; Bu nedenle, bu tedavinin sonuçlarını daha net bir şekilde
tanımlamak için ek araştırmalara ihtiyaç vardır.
Genel olarak, hastalığın
tedavisi semptomatik ve destekleyicidir. Ağrı tedavisine yardımcı olmak
için ilaçlar verilebilir ve fizik tedavi kas tonusunu ve dolaşımını sürdürmeye
veya artırmaya yardımcı olabilir.
Adie (1926) ilk önce narkolepsiyi ayrı ve
spesifik bir varlık olarak tanımladı. Gündüz uyuşukluğunu ve düşük uyanıklığı
engelleme saldırılarıyla karakterize bir uyku bozukluğudur. Derin uyku evresi
(REM) uykusunun normal fizyolojik bileşenleri, rüya görme ve kas tonusu kaybı
ayrılır ve aynı zamanda özne uyanıkken de ortaya çıkar ve yarı uyku rüyalar ve
iskelet kası felci ve atoni bölümleri (katapleks ve uyku felci) ile sonuçlanır.
). Normal uykunun aksine, narkolepsinin sıklığı genellikle REM aktivitesiyle başlar
ve uykuya dalma süresi normalden kısadır.Etkilenen bireyler gün boyunca yorgun
hissederler ve günde birkaç kez uyumak için çok zorlayıcı bir dürtü
yaşayabilirler. “Uyku saldırıları”, alışılmadık zamanlarda, örneğin yemek
sırasında veya bir konuşmanın ortasında olabilir. Birkaç saniyeden birkaç
dakikaya kadar dayanır ve sıklıkla daha uzun bir uykuya yol açar.
Narkolepsinin bir diğer ortak özelliği de,
güçlü duygulara (anlar, gülme, sürpriz veya öfke gibi) tepki olarak ani bir kas
tonusu kaybı olan katapleksidir. Bu kas zayıflığı bölümleri etkilenen bir
kişinin çökmesine veya düşmesine neden olabilir, bu da zaman zaman
yaralanmalara neden olabilir. Katapleksi bölümleri genellikle birkaç saniye
sürer ve bunlar günde birkaç kez ila yılda birkaç kez meydana gelebilir.
Narkolepsi tanısı alan çoğu insanda da katapleksi vardır. Bununla birlikte,
bazıları, araştırmacıları durumun iki ana biçimini ayırt etmeye
yönlendirmiştir: katapleksi olan narkolepsi ve katapleksi olmayan narkolepsi.
Görülme Sıklığı
Narkolepsi, Amerika Birleşik Devletleri ve
Batı Avrupa’daki yaklaşık 2,000 kişiden 1’ini etkiler. Bununla birlikte,
hastalığın, özellikle hafif semptomları olan kişilerde yetersiz teşhis edilmesi
muhtemeldir. Dünya çapında, narkolepsi en fazla 600 kişiden 1’ini etkilediği
Japonya’da görülüyor.
Oluşum Nedeni
Narkolepsi muhtemelen bazıları tanımlanmış
fakat çoğu bilinmeyen kalmaya devam eden genetik ve çevresel faktörlerin bir
kombinasyonundan kaynaklanmaktadır.
Çoğu katapleksi olan narkolepsi
vakalarında ve bazılarında katapleksi olmayan vakalarda uyku anormallikleri,
beynin bir kısmındaki hipotalamus adı verilen belirli beyin hücrelerinin
(nöronlar) kaybından kaynaklanır. Bu hücreler normalde, vücutta birçok önemli
işlevi olan hipokretinler (ayrıca oreksin olarak da bilinir) denilen
kimyasalları üretir. Özellikle, hipokretinler günlük uyku-uyanıklık döngüsünü
düzenler. Narkolepsili kişilerde hipokretin üreten nöronların ölümünü neyin
tetiklediği açık değildir, ancak kanıtlar giderek artan bir şekilde bağışıklık
sisteminin anormalliğine işaret etmektedir.
Araştırmacılar çeşitli genlerde narkolepsi
gelişme riskini etkileyen değişiklikler tespit etmişlerdir. Bu genlerin en iyi
araştırılanı, bağışıklık sisteminde önemli bir rol oynayan bir proteinin bir
parçasını yapmak için talimatlar veren HLA-DQB1’dir. HLA-DQB1 geni, insan
lökosit antijeni (HLA) kompleksi olarak adlandırılan gen ailesinin bir
parçasıdır. HLA kompleksi, bağışıklık sisteminin vücudun kendi proteinlerini
yabancı istilacıların (virüsler ve bakteriler gibi) ürettiği proteinlerden
ayırmasına yardımcı olur. HLA-DQB1 geni, her bir kişinin bağışıklık sisteminin
çok çeşitli yabancı proteinlere reaksiyona girmesine izin veren birçok farklı
normal varyasyona sahiptir. HLA-DQB1 * 06: 02 olarak adlandırılan HLA-DQB1
geninin bir varyasyonu, özellikle katapleksi ve hipokretin kaybı olan kişilerde
narkolepsi ile güçlü bir şekilde ilişkilendirilmiştir. Narkolepsili çoğu insan
HLA-DQB1 * 06: 02 varyasyonuna sahiptir ve birçoğunun, birbiriyle yakından
ilişkili HLA genlerinin spesifik versiyonları vardır. Bu genetik
değişikliklerin durumun gelişme riskini nasıl etkilediği açık değildir.
Bazı ek genlerdeki varyasyonlar
narkolepsiyle de ilişkilendirilmiştir. Bu genlerin çoğunun bağışıklık sistemi
fonksiyonunda rol oynadığı düşünülmektedir. Bununla birlikte, bu genlerdeki
değişiklikler muhtemelen narkolepsi geliştirme riskine yalnızca küçük bir katkı
yapar. Diğer genetik ve çevresel faktörlerin de bir kişinin bu hastalığı
geliştirme şansını etkilemesi muhtemeldir. Örneğin, çalışmalar strep boğaz
(streptococcus), soğuk algınlığı ve grip gibi bakteriyel veya viral
enfeksiyonların risk altındaki insanlarda narkolepsiyi tetiklemede rol
oynayabileceğini göstermektedir.
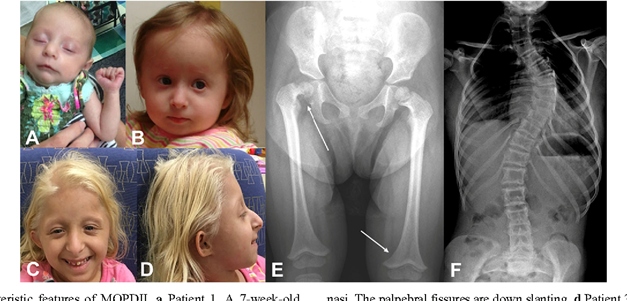
MOPD2, kısa boyluluk (cücelik); iskelet anormallikleriyle, anormal küçük
kafa yapısına (mikrosefali) sahip olmayla ve diş çıkarmada anormalliklerle
ilişkilendirilmiş nadir bir hastalıktır. Büyüme problemleri doğumdan önce
görülmeye başlayabilir ve doğum öncesinde bebeğin büyümesi de yavaştır. Doğumdan
sonra da büyümesinde yavaşlık görülür. Yetişkin bir bireyin beyin boyutu, 3
yaşındaki bir çocuğun beyninin boyutuna eş değerdir ama zekâ gelişiminde bir
gerilik yoktur. Bu hastalığa sahip bir yetişkinin boyu 50,8 -101,6 cm arası değişmektedir.
Genetik Değişiklikler/Etken Faktörler
PCNT
genindeki mutasyonlar MOPD 2’ye neden olmaktadır. Bu gen, pericentrin adlı
proteinin yapımında rol oynar. Bu protein, hücrelerde sentrozomların içinde
bulunmaktadır. Pericentrin, diğer proteinlerin sentrozomda sağlamlaşmasına
yardım etmektedir böylece diğer proteinlerle etkileşim içinde olup hücre
döngüsünde rol oynar. PCNT genindeki mutasyonlar fonksiyonu bozulmuş pericentrin
proteinin üretilmesine sebep olur bu da diğer proteinlerin sentrozoma sabitlenmelerini
engeller. Böylece sentrozomlar, mikrotübülleri doğru bir şekilde bir araya
toplayamaz ve bu da hücre döngüsünü ve hücre bölünmesini etkiler. Hücre
bölünmesinin bozulması hücre üretilmesinin azalmasına sebep olur. Toplam hücre
sayısının az olması kemiklerin kısa olmasına ve MOPD2’nin diğer belirti ve semptomlarına
sebep olur
Belirti ve Semptomlar
Kalça çıkığı; kol ve bacak kemiklerinin incelmesi; kemiğin son kısmının
şeklinin anormal olması; kemiğin uzayan geniş kısmında anormallik; küçük
el ve ayak parmakları; serçe parmağının eğri olması; omurga eğriliği; el bilek kemiklerinin kısalığı; çok
tiz ses; karakteristik yüz özellikleri (belirgin burun, dolu yanaklar, yüzün
orta kısmının uzun olması ve küçük çene); küçük dişler; küçük kulak lobları; anormal
deri pigmentasyonu ve kan damarlarında anormallikler (beynin merkezindeki kan
damarında şişlik meydana gelebilir ve bu da beyinde kanamaya sebep olabilir) .
Genetik Görülme Sıklığı
Bu hastalık çok nadir
olduğu için yaygınlığı belirsizdir.
Kalıtım Paterni/Deseni
Otozomal resesif.
Teşhis Yöntemleri ve Tedaviler
MOPD 2
genetik bir nadir hastalık olduğu için tedavisi bilinmemektedir. Teşhis için genetik
testi yapılması önerilir.
Hastalıkla İlişkili Genler
PCNT
genindeki mutasyonlar.
Hastalığın Diğer İsimleri
MOPD 2; MOPD II; Osteodysplastic primordial dwarfism type 2
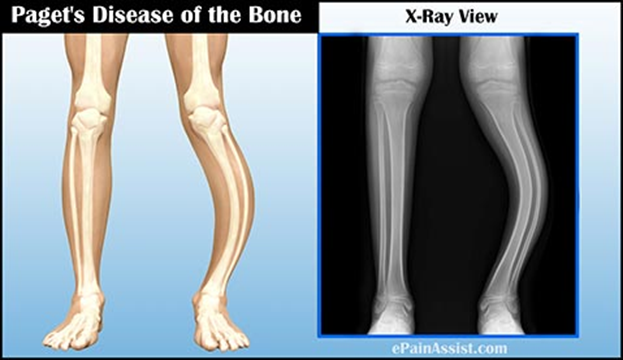
Kemiğin Paget hastalığı, kemiklerin
normalden daha büyük ve daha zayıf büyümesine neden olan bir hastalıktır.
Etkilenen kemikler şekilsiz ve kolay kırılabilir.
Klasik kemik Paget hastalığı şekli tipik
olarak orta yaşta veya daha sonra ortaya çıkar. Genellikle bir veya birkaç
kemikte meydana gelir ve bir kemikten diğerine yayılmaz. Her ne kadar kemik,
pelvis, kafatası veya bacaklardaki kemikleri etkilese de, herhangi bir kemik de
etkilenebilir.
Klasik Paget kemiği hastalığına sahip
birçok kişi, kemik anormallikleri ile ilişkili herhangi bir semptom yaşamaz.
Hastalık genellikle beklenmedik şekilde röntgen veya başka nedenlerle yapılan
laboratuar testleri ile teşhis edilir. Semptom geliştiren kişilerin acı çekmesi
daha olasıdır. Etkilenen kemiklerin kendileri ağrılı olabilir veya yakın
eklemlerdeki artrit nedeniyle ağrı oluşabilir. Artrit, kemiklerin, özellikle de
bacaklardaki ağırlık taşıyan kemiklerin distorsiyonunun, eklemlerde ekstra
aşınma ve yıpranmaya neden olmasıyla sonuçlanır. Artrit, en sık bu hastalığı
olan kişilerde dizleri ve kalçaları etkiler.
Paget kemiği hastalığının diğer
komplikasyonları hangi kemiklerin etkilendiğine bağlıdır. Hastalık kafatasının
kemiklerinde ortaya çıkarsa, genişletilmiş bir kafa, işitme kaybı, baş ağrısı
ve baş dönmesine neden olabilir. Hastalık omurgadaki kemikleri etkilerse, uyuşukluğa
ve karıncalanmaya (sıkışan sinirler nedeniyle) ve anormal omurga eğriliğine yol
açabilir. Bacak kemiklerinde, hastalık eğri bacaklara ve yürüme zorluğuna neden
olabilir.
Osteosarkom adı verilen nadir bir kemik
kanseri türü, Paget kemiği hastalığı ile ilişkilendirilmiştir. Bu tip kanser
muhtemelen bu hastalığa sahip 1000 kişiden 1’inden azında ortaya çıkar.
Görülme Sıklığı
Klasik kemik kemiği hastalığı, Amerika
Birleşik Devletleri’nde 40 yaşından büyük kişilerin yaklaşık yüzde 1’inde
görülür. Bilim adamları bu ülkede yaklaşık 1 milyon insanın hastalığa sahip
olduğunu tahmin ediyorlar. Batı Avrupa mirası insanlarında en yaygın olanıdır.
Erken başlayan kemik Paget hastalığı çok
daha nadirdir. Hastalığın bu formu yalnızca birkaç ailede bildirilmiştir.
Hastalığa Sebep Olan Etmenler/Etkenler
Genetik ve çevresel faktörlerin bir
kombinasyonu muhtemelen kemiğin Paget hastalığına neden olmada rol oynar.
Araştırmacılar, birkaç gende bozukluk riskini artıran değişiklikler tespit
etmişlerdir. Belirli virüslere sahip enfeksiyonlar dahil olmak üzere diğer
faktörler, risk altındaki kişilerde hastalığın tetiklenmesinde rol oynayabilir.
Ancak, genetik ve çevresel faktörlerin kemiğin Paget hastalığının gelişimi
üzerindeki etkisi belirsizliğini koruyor.
SQSTM1, TNFRSF11A ve TNFRSF11B genleri,
kemik düzenlenmesinde rol oynar(Bone Remodelling); bu, eski kemiğin
parçalandığı ve yerine yeni kemiğin oluşturulduğu normal bir işlemdir. Kemikler
sürekli yenileniyor ve kemiklerin güçlü ve sağlıklı kalmasını sağlamak için
süreç dikkatle kontrol ediliyor. Kemiğin Paget hastalığı kemik yeniden
şekillenme işlemini bozar. Etkilenen kemik anormal şekilde parçalanır ve
normalden çok daha hızlı bir şekilde değiştirilir. Yeni kemik dokusu büyüyünce
normal kemikten daha büyük, daha zayıf ve daha az organize olur. Kemik tadilatı
ile ilgili bu sorunların neden bazı hastalıkları etkilediği, ancak bu hastalığa
sahip insanlarda diğerlerini etkilemediği açık değildir.
Araştırmacılar, bir insanın Paget
hastalığı geliştirme şansını etkileyebilecek ilave genler aramaktadır.
Çalışmalar, kromozom 2, kromozom 5 ve kromozom 10’un belirli bölgelerindeki
genetik çeşitlemelerin hastalık riskine katkıda bulunduğunu göstermektedir.
Bununla birlikte, bu kromozomlar üzerindeki ilişkili genler tanımlanmamıştır.
Tedavi
Paget hastalığı olan hasta için dört ana
tedavi yöntemi vardır: farmakolojik olmayan terapi (temel olarak bazı ağrı
tiplerini kontrol etmeye yardımcı olmak için kas gücünü arttırmanın bir aracı
olarak fiziksel tedaviye odaklanma); bifosfonatlar veya kalsitoninler
kullanarak farmakolojik tedavi; analjezik kullanarak ağrı yönetimi; ve cerrahi
müdahale.
Pharmacological Treatment
Bisfosfonatlar osteoklastlarla kemik
emilimini baskılar veya azaltır. Bunu hem doğrudan, osteoklastların işe alımını
ve fonksiyonunu engelleyerek hem de dolaylı olarak osteoblastların bir
osteoklast formasyonu inhibitörü üretmek için uyararak yaparlar. Artık bu
ilaçların nasıl çalıştığı konusunda makul bir anlayış var ve çeşitli bifosfonat
türleri arasındaki farklar daha iyi anlaşılıyor.
Ağrı Yönetimi: Analjezikler
Doğrudan Paget hastalığına atfedilebilen
ağrı genellikle yukarıda tarif edildiği gibi anti-osteoklast tedavisi ile
hafifletilir. Bazı ağrı kemik deformitesinin veya artritik veya nörolojik
komplikasyonların sonucu olabilir. Bu durumda, asetaminofen, steroidal olmayan
antienflamatuar ilaçlar (NSAIDS) veya cox-2 inhibitörleri, seçilen ana pagetik
tedaviye ek olarak, pagetik ağrının tedavisinde faydalı olabilir.
Kalsitoninler
Paget
hastalığı için deri altına salmon kalsitonin enjeksiyonu ilk kez uygulanan
tedavi olmuştur. Somon kalsitonininin kemik dönüşüm endekslerini yüzde 50
oranında azalttığı, kemik ağrısı semptomlarını azalttığı, etkilenen kemiklerin
sıcaklığını azalttığı, bazı nörolojik komplikasyonları iyileştirdiği ve litik
lezyonların iyileşmesini sağladığı gösterilmiştir. Günümüzde kullanımı
çoğunlukla bifosfonatları tolere etmeyen hastalarla sınırlıdır. Somon
kalsitonine karşı ikincil direnç durumunda, bifosfonat tedavisine geçiş
esastır. Haftada üç kez veya günde üç kez yapılan bir enjeksiyon olan
Miacalcin, Paget hastalığı için onaylanan tek kalsitonindir.
Zoledronik asit (Reclast) tercih edilen tedavidir.
İntravenöz uygulama 15 dakika zarfında
verilen 5 mg infüzyondadır. İnfüzyon sonrası düşük kan kalsiyum riskini
azaltmak için hastalara ayrıca iki hafta boyunca günde 1500mg birim kalsiyum ve
1000 birim D vitamini almalıdır. Zoledronik asit (Reclast) düşük kan kalsiyum
veya D vitamini eksikliği olan kişiler ve böbrek fonksiyon bozukluğu olan
kişiler için endike değildir.
Zoledronic acid (Reclast), Paget
hastalığının Aclasta adı altında Amerika Birleşik Devletleri dışındaki bazı
ülkelerde de tedavi edilmesi için pazarlanmaktadır.
Hafif bir Paget hastalığı şekli, bir veya
iki 60 mg pamidronat disodyum infüzyonu ile bastırılabilir, oysa hastalığın
daha ağır bir şekilde ortaya çıkması, haftada bir veya iki haftada birkaç kez
60-90 mg pamidronat infüzyonu gerektirebilir. Serum alkalin fosfataz testi,
uygun miktarda infüzyonun uygulanmasından yaklaşık iki ila üç ay sonra
yapılmalıdır. Pamidronat disodyumun birkaç genel formu da mevcuttur.
Hipokalsemiyi azaltmak için bu tedaviyi
kullanan ve ortak bir yan etki olan hastalar için oral kalsiyum ve D vitamini
desteği önerilmektedir ve düşük kan kalsiyum düzeyi ve D vitamini eksikliği
olan hastalar ödenen disodyum ile tedavi edilmemelidir.
Düşük kan kalsiyum veya D vitamini
eksikliği olan hastalar zoledronik asit ile tedavi edilmemelidir ve böbrek
fonksiyonlarını tehlikeye atan hastalar için ilaç endike değildir.
Refsum
hastalığı görme kaybına,koku duyusunun
yokluğuna(anosmi) ve diğer çeşitli işaret ve semptomlara sebep olan kalıtımsal
bir durumdur.
Refsum hastalığıyla
ilişkili görme kaybı retinitis pigmentosa denilen göz hastalığı sebebiyle
olur.Bu hastalık gözün arka tarafında ışığa hassas katmanı yani retinayı
etkiler.Görme kaybı retinanın ışığa duyarlı hücrelerinin gitgide kötüleşmesiyle
meydana gelir.Retinis pigmenosanın ilk işareti genellikle çocuklukta ortaya
çıkan gece görüşünün kaybıdır.Yıllar ilerledikçe,hastalık taraflı(periferal)
görüşü bozar ve nihayetinde körlüğe yönlendirebilir.
Görme kaybı
ve anosmi Refsum hastası olan neredeyse herkeste görülür ama diğer işaretler ve
semptomlar çeşitlidir.Etkilenmiş bireylerin yaklaşık bir-üçü ellerde ve ayaklarda anormalliklerle
doğarlar.Daha sonra hayatta ortaya çıkan özellikler ilerleyici kas güçsüzlüğü
ve kaybı;kötü denge ve koordinasyon(ataksi);duyma kaybı;ve kuru,pullu
deridir(balık pulu hastalığı).Ek olarak Refsum hastalığına sahip bazı
insanlarda anormal kalp ritmi(aritmi) ve buna bağlı olarak kalp problemleri
gelişir ve bu hayatı tehdit edici olabilir.
Sıklık
Refsum hastalığının prevelansı bilinmemesine rağmen nadir bir
durum olduğu düşüncesi vardır.
Sebepleri:
Olguların %90 dan fazlasında PHYH genindeki mutasyonlar
sonucunda Refsum hastalığı oluşur.Arta kalan olgularda PEX7 denen genin
mutasyonları hastalığa sebep olur.
Refsum hastalığının işaret ve semptomları fitanik asi denen
bir tip yağ asidinin anormal şekilde üretilmesinin sonucu olarak oluşur.Bu
madde özellikle sığır eti ve süt ürünleriyle beslenmeyle elde edilir.Genelde alfa
oksidasyon olarak adlandırılan süreç vasıtasıyla yıkılmasıyla hücrenin içinde
peroksizom denen yapılar ortaya çıkar.Bu kese benzeri kompartımanlar yağ
asitleri ve belirli toksik bileşikler bir çok farklı maddeyi işleyen enzimleri
içerir.
PHYH yada PEX7 geninin mutasyonlarının her iki durumunda da
fitanik asitin yıkımını içeren
peroksizomların olağan fonksiyonlarını aksatır.Sonuç olarak bu madde vücut
dokularında birikir.Fitanik asitin birikimi,bu maddenin aşırı birikmesinin
görme,koklama ve Refsum hastalığının diğer spesifik özelliklerine sebep olması
belirsiz olmasına rağmen hücreler için toksiktir.
İşaret ve Semptomlar
Refsum hastalığının semptomları görme harabiyeti,dejeneratif
sinir hastalığı,kas koordinasyonunun kaybı,kemik ve deri değişikliklerini
içerir.Semptomlar gece körlüğünü,periferal görüşün kaybını ve kas
koordinasyonunun kaybıyla ilişkili olarak hissizlik ve güçsüzlüğü içerebilir.
Etkilenmiş bireyler kollarda ve bacaklarda olağandışı yanma
ve karıncalanma hissi(parestezi) deneyimleyebilir.Nörolojik semptomlar sık
düşmelerle birlikte istikrarsız yürüyüş(ataksi) ve periferal nöropatiyi(sensör,motor
ve refleks değişiklikleriyle karakterize) içerir.Deri değişiklikleri
kuruluk,kaşıntı ve pullanmayı içerir.
Refsum
hastalığının infantil formunda var olması
genellikle hayatın ilk yılında ortaya çıkmasıyla birliktedir.Erken
başlangıca ek olarak,görme ve duymanın harabiyeti,karaciğerde büyüme ve safra
asitinin yetersiz metabolizmasıyla ve
gelişmenin gecikmesiyle karakterizedir.
Refsum Hastalığının Diğer İsimleri
Yetişkin Refsum Hastalığı
ARD
CRD
HMSN 4
HMSN tip 4
DOC
11(Fitanik Asit Tip)
Klasik Refsum Hastalığı
Kalıtsal Motor ve Duyusal Nöropati
Tip 4
Heredopati ataktik polinöritiformis
Fitanik Asit Depolama Hastalığı
Refsum Sendromu
Refsumun Hipertrofik Nöropatisi
Boynuzlaşma 11’in Hastalığı(Fitanik
Asit Tip)
Etkilenmiş Popülasyonlar
Refsum hastalığının başlangıç yaşı fazlasıyla çeşitlilik
göstermektedir.Erken çocukluktan 50 yaşına kadar herhangi bir yaşta meydana
gelebilir ama semptomlar çoğunlukla 20 yaşında görünür.Erkekler ve kadınlar
eşit sayılarda etkilenir.
Kalıtım Paterni
Bu durumun kuşaktan kuşağa aktarılması her hücredeki genin
iki kopyasında da mutasyona sahip olması anlamına gelen otozomal resesif
paternle olur.Otozomal resesif durumlu bireyin ebeveynleri mutasyonlu genden
birer kopya taşırlar ama tipik olarak durumun işaret ve semptomlarını
göstermezler.
Refsum hastalığından sorumlu genler kromozom 10’un kısa
kolunda ve kromozom 6’nın uzun kolunda bulunur.
Teşhis
Teşhiste fitanik asitin mevcudiyeti kanda yada idrar
örneklerindedir.
Standart Terapiler
Tedavi
Refsum’un
tedavisi fitanik asiti düşük(süt ürünlerinde,sığır etinde,kuzuda ve bazı deniz
ürünlerinde bulunur) ve kalorisi yüksek
sıkı bir diyeti takip etmeyi kapsar.Kan plazmasının ortadan kaldırılması ve
reinfüzyonuna(plazmaferez) ayrıca gerek duyulabilir.Diğer tedavi semptomatik ve
destekleyicidir.
Mukopolisakaridozlar (MPS rahatsızlıkları), mukopolisakaritler adı verilen karmaşık şeker moleküllerini parçalamak için gereken enzimlerden birinin eksikliğinden kaynaklanan yedi nadir genetik hastalık grubudur. Mukopolisakaritler bazen GAG (glikoaminoglikanlar) olarak da adlandırılır ve vücut tarafından doğal olarak üretilir ve kemik, kıkırdak, cilt ve dokuların yapımında kullanılır. Mukopolisakaritler, isimlerini, kalın jöle benzer kıvamlarından (“muko”) alır; “poli”, birçok anlam ifade eder ve “sakarit” anlamına gelir. Beyin de dahil olmak üzere merkezi sinir sistemi hücrelerinde yüksek konsantrasyonlarda mukopolisakarit, bu bozukluklara eşlik eden nörolojik ve gelişimsel sorunlara neden olur. MPS bozuklukları, “lizozomal depo bozuklukları” olarak bilinen daha geniş bir hastalık grubunun parçasıdır çünkü mukopolisakkaritleri parçalayan enzimler, lizozom adı verilen hücrenin bir bölümünde bulunur. Lizozom, sıklıkla hücrenin “atık imha tesisi” olarak adlandırılır. 50’den fazla lizozomal depo bozukluğu vardır. MPS III (Sanfilippo sendromu), heparan sülfat adı verilen belirli bir mukopolisakkariti parçalayabilen enzim eksikliğinden kaynaklanır. Sanfilippo sendromu olarak da bilinen mukopolisakkaridoz tip III (MPS III), öncelikle beyni ve omuriliği etkileyen merkezi bir sistemdir (merkezi sinir sistemi). Diğer vücut sistemleri de dahil edilebilir.MPS III’lü kişiler genellikle doğumda durumun hiçbir özelliğini göstermezler, ancak erken çocukluk döneminde hastalığın belirtileri ve semptomlarını göstermeye başlarlar. Etkilenen çocuklar genellikle başlangıçta gecikmiş konuşma ve davranış problemleri yaşarlar. Huzursuz, yıkıcı, endişeli veya saldırgan olabilirler ve sosyal etkileşim ve iletişim zorluğu ile karakterize bir durum olan otizm spektrum bozukluğunun bazı özelliklerini sergilerler. MPS III’lü çocuklarda uyku bozuklukları da çok yaygındır. Bu durum ilerleyici zihinsel yetersizliğe ve daha önce edinilmiş becerilerin kaybına neden olur (gelişimsel regresyon). Hastalığın ilerleyen aşamalarında, MPS III’lü kişiler nöbet ve hareket bozuklukları geliştirebilir. MPS III’ün fiziksel özellikleri diğer mukopolisakkaridoz tiplerinden daha az belirgindir. MPS III’lü bireyler tipik olarak hafif “kaba” yüz özelliklerine, büyük bir kafaya (makrosefali), hafifçe genişletilmiş bir karaciğere (hafif hepatomegali) ve göbek deliğinin (göbek fıtığı) veya alt karnının (kasık fıtığı) etrafındaki yumuşak bir dış torbaya sahiptir. ). MPS III’lü bazı kişilerde kısa boy, eklem sertliği veya hafif disostoz multipleks vardır, bu da röntgende görülen çok sayıda iskelet anomalisine karşılık gelir. Etkilenen bireyler genellikle kronik ishal ve tekrarlayan üst solunum ve kulak enfeksiyonları yaşarlar. MPS III’lü kişilerde ayrıca işitme kaybı ve görme sorunu olabilir. MPS III, genetik nedenleriyle ayırt edilen IIIA, IIIB, IIIC ve IIID tiplerine ayrılmıştır. Farklı MPS III tipleri benzer belirti ve semptomlara sahiptir, ancak MPS IIIA’nın özellikleri tipik olarak yaşamda daha erken ortaya çıkmakta ve daha hızlı ilerlemektedir. MPS III’lü insanlar genellikle ergenlik veya erken yetişkinlik döneminde yaşarlar.
Şekil.1. Sanfilippo Hastası Bir Birey
Genetik Değişiklikler ve Etken
Faktörler
GNS,
HGSNAT, NAGLU ve SGSH genlerindeki mutasyonlar MPS III’e neden olur. Bu genler,
glikosaminoglikanlar (GAG’ler) adı verilen büyük şeker moleküllerinin
parçalanmasında rol oynayan enzimlerin hazırlanmasına yönelik talimatlar
sağlar. GNS, HGSNAT, NAGLU ve SGSH enzimleri, heparan sülfat adı verilen bir
GAG alt kümesinin kademeli olarak parçalanmasında rol oynar.
MPS IIIA, SGSH genindeki mutasyonlardan, MPS IIIB ise NAGLU gen mutasyonlarından
kaynaklanır. HGSNAT genindeki mutasyonlar MPS IIIC ile sonuçlanır ve GNS gen
mutasyonları MPS IIID’ye neden olur. Bu genlerdeki mutasyonlar enzim
fonksiyonunu azaltır veya yok eder. Bu enzimlerden herhangi birinin eksikliği,
heparan sülfatın parçalanmasını bozar. Sonuç olarak, kısmen parçalanmış heparan
sülfat hücreler içerisinde, özellikle lizozomların içinde birikir. Lizozomlar,
farklı molekül türlerini sindiren ve geri dönüştüren hücrede bulunan
bölümlerdir. Moleküllerin lizozomlar içinde birikmesine neden olan MPS III gibi
durumlara lizozomal depo bozuklukları denir. Araştırmacılar, GAG birikiminin
lizozomların içindeki diğer proteinlerin fonksiyonlarına müdahale ettiğine ve
hücrelerin normal fonksiyonlarını bozduğuna inanmaktadır. Heparan sülfat
oluşumunun neden MPS III’te merkezi sinir sistemini etkilediği bilinmemektedir.
Belirti ve Semptomlar
Sanfilippo
sendromu (MPS Tip III) olan hastalar genellikle doğumda normal görünür, ancak
gelişimsel gecikme genellikle 2-5 yaş arasında belirgindir. Zihinsel ve motor
gelişim 3-6 yaşları arasında bir zirveye ulaşır ve sonrasında davranış
bozuklukları ve entelektüel düşüş genellikle meydana gelir. Bununla birlikte,
hiperaktivite ve huzursuzluk gibi davranışsal sorunlar daha önce
belirginleşebilir. Şiddetli davranış bozuklukları, Sanfilippo sendromunun çok
yaygın bir özelliğidir ve hastalığın yönetimi zor olan yanlarından biridir. Diğer
semptomlar kaba saç, aşırı saç büyümesi (hirsutizm), hafif kaba yüz
özellikleri, uyku problemleri, hafifçe genişlemiş karaciğer ve / veya dalak,
konuşma gecikmesi, solunum ve kulak enfeksiyonları, ishal, fıtıklar, nöbetler
ve titrek ve düzensiz bir yürüyüş olabilir . Bazı çocuklarda işitme kaybı ve
görme bozukluğu gelişebilir. Sanfilippo sendromu olan çocuklar genellikle
entelektüel fonksiyonlarını yitirmeye başlar, özellikle motor fonksiyonlar
düşmeden önce konuşma. Bazılarında hızlı fonksiyon kaybı, bazılarında ise
hastalığın çok daha yavaş ilerleyişini gösteren kardeşler arasında bile,
regresyon oranında büyük farklılıklar vardır. Ölüm, 10 yaşından önce, yaşamın
üçüncü veya dördüncü yıllarına kadar, ortalama 15 ile 20 yaşlarında olabilir.
Genetik Görülme Sıklığı
MPS
III, görülme sıklığı 100.000 doğumda 0.28 ile 4.1 arasında olduğu bildirilen
nadir bir hastalık olarak sınıflandırılır. MPS IIIA, 100.000 doğumda 1’i
etkileyen en yaygın alt tiptir, bunu 200.000’de 1’de B tipi takip eder. Güney
Avrupa’daki bazı ülkelerde, B tipi tipinin A’dan daha yaygın olduğu
bildirilmiştir. MPS IIIC ve IIID, sırasıyla 1,5 milyonda 1 ve 1 milyon doğumda
1 oranında görülme sıklığı ile nadirdir.
Kalıtım Paterni/Deseni
Dört
MPS III tipinin hepsine, heparan sülfatı parçalayan enzimlerin hazırlanmasına
yönelik talimatlar içeren farklı genlerdeki değişiklikler (mutasyonlar) neden
olur.
TİP
GEN
ENZİM
MPS IIIA
SGSH
Heparan N-sülfataz
MPS IIIB
NAGLU
a-N-asetilglukosaminidaz
MPS IIIC
HGSNAT
Heparan-alfa-glukosaminit N-Asetil transferaz
MPS IIID
GNS
N-asetilglukozamin 6-sülfataz
MPS
III otozomal resesif bir şekilde kalıtsaldır. Bu, her iki ebeveynin de değişmiş
genin bir kopyasına ve bir normal kopyasına sahip olduğu anlamına gelir –
taşıyıcılar olarak bilinir ve durumun belirtilerini göstermezler. MPS III’e
sahip bir çocuk, değiştirilmiş genin iki kopyasını, her bir ebeveynden bir tane
alır.
Otozomal
resesif kalıtımda, her iki taşıyıcıya sahip bir çiftin her bir hamileliğinde:
-%
25 (4’te 1) etkilenen bir çocuğa sahip olma şansı
-%
50 (2’de 1) çocuğun değişmiş genin sadece bir kopyasını alma ve dolayısıyla
taşıyıcı olma şansı
-%
25 (4’te 1), bir çocuğun ne etkileneceğini ne de taşıyıcıyı etkileme şansı.
Risk
erkeklerde ve kadınlarda aynıdır.
Teşhis Yöntemleri ve Tedaviler
MPS
III’ü teşhis etmek için, genellikle mukopolisakaritler (GAG’ler) ilk önce
idrarda ölçülür, ardından kandaki enzim aktivitesinin veya küçük bir cilt
numunesinin ölçümü yapılır. İdrarda artan heparan sülfat ve dört enzimden
herhangi birinin aktivitesindeki bir azalma (yukarıdaki tabloda gösterilmiştir)
genellikle MPS III teşhisi ile tutarlıdır ve MPS III tipini (A, B, C veya D)
tanımlayacaktır. ). MPS III tipinin bilinmesi önemlidir, çünkü bunun genetik
testi kolaylaştıracağı ve daha da önemlisi, geliştirilen tedavilerin çoğu
sadece spesifik tipler içindir. Bir kan numunesinin genetik testi, DNA’daki
kesin değişikliklerin tanımlanmasına izin verecektir. Ailedeki diğer
çocukların, gelecekteki gebeliklerin ve geniş aile üyelerinin etkilerini
öğrenmek için genetik danışmanlığa katılmak önemlidir. Danışman kalıtım
modelini açıklayacak ve kimin test edilmesi gerektiği konusunda tavsiyede
bulunacaktır.Genetik tanı biliniyorsa, bu bilgi ailenin risk altındaki diğer
üyelerini test etmek için kullanılabilir. Ayrıca gelecekteki gebeliklerin doğum
öncesi testleri (hala anne karnındayken fetusun test edilmesi) ve / veya
preimplantasyon teşhisi (ilgili gen mutasyonunu taşımayanları seçmek için IVF
ile oluşturulan embriyoların test edilmesi) için kullanılabilir.
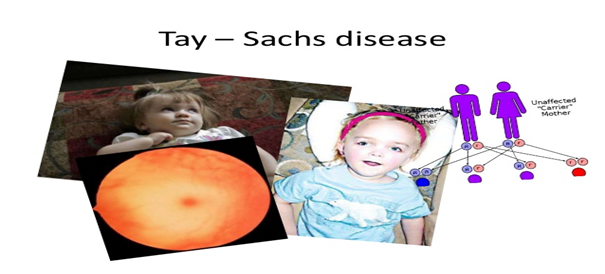
Tay-Sachs hastalığı, bir enzim eksikliğinin (hekzosaminidaz A), beyin ve sinir hücrelerinde gangliyozitler olarak bilinen bazı yağların (lipitler) aşırı birikmesine neden olduğu, nadir görülen, nörodejeneratif bir hastalıktır. Gangliosidlerin bu anormal birikimi, merkezi sinir sisteminin ilerleyici işlev bozukluğuna yol açar. Bu hastalık lizozomal depo hastalığı olarak sınıflandırılır. Lizozomlar hücrelerdeki ana sindirim üniteleridir. Lizozomlardaki enzimler, bazı kompleks karbonhidratlar ve yağlar dahil olmak üzere besin maddelerini parçalamaktadır veya “sindirmektedir”. Yağ gibi bazı maddeleri parçalamak için gerekli olan hekzosaminidaz A gibi bir enzim eksik veya etkisiz olduğunda, lizozomalde birikir. Buna “anormal depolama” denir. Lizozomda çok fazla yağ biriktiğinde hücreyi yok eden ve çevreleyen dokuya zarar veren toksik hale gelir. Tay-Sachs hastalığına bağlı semptomlar, ani seslere abartılı bir irkilme tepkisi, halsizlik, önceden edinilmiş becerilerin kaybı (yani psikomotor regresyon) ve ciddi şekilde azalmış kas tonusu (hipotoni) içerebilir. Hipotonili bebekler “disket” olarak tanımlanabilir. Hastalık ilerledikçe, etkilenen bebekler ve çocuklar gözlerin orta tabakasında kiraz kırmızısı lekeler, kademeli görme kaybı ve işitme kaybı, kas sertliği ve sınırlı hareketler (spastisite), nihai felç, kontrolsüz elektriksel rahatsızlıklar beyin (nöbet) ve bilişsel süreçlerin bozulması (demans). Tay-Sachs hastalığının klasik şekli bebeklik döneminde ortaya çıkar. Bu en yaygın şeklidir ve genellikle erken çocukluk döneminde ölümcüldür. Tay-Sachs hastalığının genç ve yetişkin formları da vardır, ancak bunlar nadirdir. Çocuk formuna sahip, alt form olarak da adlandırılan çocuklar, çocuk formuna sahip olanlardan daha sonra semptomlar geliştirir ve genellikle çocukluk veya ergenlikte yaşarlar. Geç başlangıçlı Tay-Sachs hastalığı olarak da adlandırılan yetişkin formu, ergenlik döneminden 30’lu yaşların ortalarına kadar herhangi bir zamanda oluşabilir. Belirtiler ve ciddiyet bir kişiden diğerine değişebilir. Bazı insanlar çocuk ve yetişkin formları arasında düşebilir. Tay-Sachs hastalığı, otozomal resesif bir şekilde kalıtsaldır. Hastalık, hekzosaminidaz A enziminin üretimini düzenleyen HEXA geni olarak bilinen bir genin değişmesinden (mutasyonlarından) kaynaklanır. HEXA geni, 15 kromozomunun (15q23-q24) uzun koluna (q) eşlenmiştir. Tay-Sachs hastalığının tedavisi yoktur ve tedavi, ortaya çıkan spesifik semptomları hafifletmeyi amaçlar. Tay-Sachs hastalığı için bir başka isim GM2 gangliosidosis tip 1’dir. Sandhoff hastalığı ve hekzosaminidaz aktivatörü eksikliği olarak adlandırılan, semptomlara dayanarak Tay-Sachs hastalığından ayırt edilemeyen ve sadece altta yatanları belirlemek için yapılan testlerle ayırt edilebilen diğer iki ilgili hastalık vardır. sebeb olmak. Bu iki rahatsızlık aynı zamanda heksosaminidaz aktivitesinin azalmasına neden olur, ancak farklı genlerdeki değişikliklerden kaynaklanır. Toplu olarak, bu üç hastalık GM2 gangliosidoses olarak bilinir.
Şekil.1. Tay-Sachs Hastalığı
Genetik Değişiklikler ve Etken Faktörler
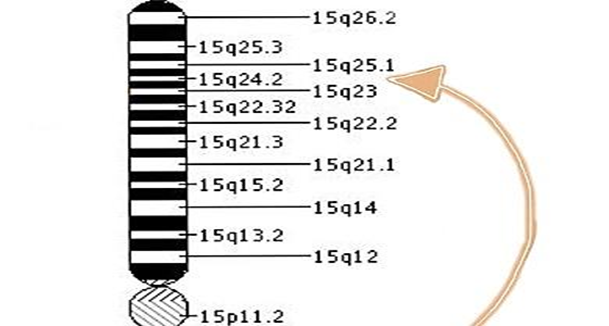
Tay-Sachs hastalığına, hekzosaminidaz alt birim alfa (HEXA) genindeki bir değişiklik (mutasyon) neden olur. Genler, vücudun birçok fonksiyonunda kritik bir rol oynayan proteinlerin oluşturulması için talimatlar sağlar. Bir genin mutasyonu meydana geldiğinde, protein ürünü hatalı, yetersiz veya eksik olabilir. Protein fonksiyonlarına bağlı olarak, bu beyin de dahil olmak üzere vücudun birçok organ sistemini etkileyebilir. HEXA geni, heksosaminidaz A enziminin üretimini düzenler. Hastalığa sahip kişilerde HEXA geninin 80’den fazla farklı mutasyonu tanımlanmıştır. HEXA geninin iki mutasyona uğramış kopyalarının (homozigotlar) miras alınması, vücut hücrelerinde GM2-gangliosid olarak bilinen yağ maddesini (lipit) parçalamak için gerekli olan hekzosaminidaz A enziminin eksikliğine neden olur. GM2-gangliyositinin parçalanamaması, beyin ve sinir hücrelerinde anormal birikimine ve sonunda merkezi sinir sisteminin ilerleyici bozulmasına neden olur. İnfantil Tay-Sachs hastalığında, neredeyse tamamen heksosaminidaz A eksikliği vardır. Geç başlangıçlı Tay-Sachs hastalığında, heksosaminidaz A enzim aktivitesinde eksiklik vardır. Bazı enzim aktivitesi olduğundan, hastalık daha az şiddetlidir ve infantil Tay-Sachs hastalığından çok daha yavaş ilerler. Geç başlangıçlı Tay-Sachs hastalığında enzim aktivitesinin tam miktarı bir kişiden diğerine büyük farklılıklar gösterir. Sonuç olarak, başlangıç yaşı, şiddeti, spesifik semptomlar ve geç başlangıçlı Tay-Sachs hastalığının ilerleme hızı da bir kişiden diğerine büyük ölçüde değişmektedir. HEXA geninde Tay-Sachs hastalığına neden olan değişiklikler otozomal resesif bir şekilde kalıtsaldır. Çoğu genetik hastalık, biri babadan diğeri de anneden alınan bir genin iki kopyasının durumuna göre belirlenir. Resesif genetik bozukluklar, bir birey aynı özellik için anormal bir genin iki kopyasını her bir ebeveynden bir tane aldığında ortaya çıkar. Bir birey hastalık için bir normal gen ve bir gen miras alırsa, kişi hastalık için taşıyıcı olur, ancak genellikle semptom göstermez. İki taşıyıcı ebeveynin hem değiştirilmiş geni geçmesi hem de etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Ebeveynler gibi taşıyıcı bir çocuk sahibi olma riski her hamilelikte% 50’dir. Bir çocuğun her iki ebeveynden normal gen alma şansı% 25’tir. Risk erkeklerde ve kadınlarda aynıdır. Araştırmacılar, Tay-Sachs hastalığı geninin, 15 (15q23-q24) kromozomunun uzun kolunda (q) bulunduğunu belirlemiştir. Kromozomlar, insan hücrelerinin çekirdeğinde bulunur ve her birey için genetik bilgiyi taşır. İnsan vücudu hücrelerinin normalde 46 kromozomu vardır. 1 ila 22 arasında numaralandırılmış insan kromozomlarının çiftlerine otosomlar denir ve cinsiyet kromozomları X ve Y olarak adlandırılır. Erkeklerde bir X ve bir Y kromozomu vardır ve dişilerde iki X kromozomu vardır. Her kromozomun “p” işaretli kısa bir kolu ve “q” işaretli uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok gruba bölünmüştür. Örneğin, “kromozom 11p13”, kromozom 11’in kısa kolundaki bant 13’ü belirtir. Numaralı bantlar, her bir kromozomda bulunan binlerce genin konumunu belirtir.
Şekil.2. Tay Sachs Hastalığı (İdeogram)
Belirti ve Semptomlar
Tay-Sachs hastalığının ilk belirtileri,
bir kişinin (varsa) ne kadar beta-heksosaminidaz A enzim aktivitesine bağlı
olarak bebeklikten yetişkinliğe kadar görülebilir. En yaygın biçimde, çocuk
formunda, bebeklerde enzim aktivitesi yoktur veya çok düşük bir seviye (%
0.1’den az) bulunur. Genellikle yenidoğan döneminde sağlıklı görünürler, ancak
3 ila 6 ay içinde semptomlar geliştirirler. İlk belirti, gürültüye karşı
abartılı bir ürkütücü tepki olabilir. Bu formdaki bebekler, yuvarlanma ve
oturma (regresyon) gibi kilometre taşlarını kaybetmeye başlar ve yavaş yavaş
felce neden olan kas zayıflığı geliştirir. Ayrıca zihinsel işlevlerini
kaybederler ve çevrelerine karşı giderek daha tepkisiz hale gelirler. 12
aylıkken, daha hızlı bir şekilde bozulmaya başlar, körlük, tedavisi zor olan
nöbetler ve yutkunma zorlaşır. Bu Tay-Sachs hastalığına sahip bebekler tipik
olarak 4 yaşından sonra hayatta kalamazlar. En sık ölüm nedeni akciğer iltihabından
(bronkopnömoni) gelen komplikasyonlardır. Çocuk formu daha az yaygındır ve
tipik olarak normal aktivitenin% 1’inden daha az enzim aktivitesine sahip
olması ile karakterize edilir. Tam olarak ne kadar aktivite olduğuna bağlı
olarak, belirtiler çocukluk döneminde, en sık 2 ve 5 yaşları arasında herhangi
bir zamanda başlayabilir. Bu forma sahip çocuklar sıklıkla sık enfeksiyonlar,
davranışsal problemler geliştirir ve yavaş yavaş ilerleyen hareket kontrolü,
konuşma ve Zihinsel işlev. Ayrıca nöbet geçirmeye başlayabilir ve görüşlerini
kaybedebilirler. Çocuk formuna sahip çocuklar, geç çocukluk veya ergenlik
döneminden vefat etmeden önce, genellikle birkaç yıl boyunca hiçbir yanıt veya
farkındalığa sahip değillerdir. Enfeksiyon yaygın bir ölüm nedenidir. Bazen
yetişkin veya kronik form olarak adlandırılan geç başlangıç formu da daha az
yaygındır ve normal enzim aktivitesinin% 10’undan azına sahip olması ile
karakterize edilir. Belirtiler ve ciddiyet, bu forma sahip kişiler arasında
daha fazla değişiklik gösterir. Belirtiler çocuklukta yetişkinliğe kadar
başlayabilir, ancak hastalık ergenliğe veya yetişkinliğe kadar sıklıkla teşhis
edilmez. Nörolojik bozukluk yavaş yavaş ilericidir ve sakarlığa ve koordinasyon
kaybına, kas güçsüzlüğü, titreme, konuşma zorluğu veya yutkunma ve kontrol
edilemeyen kas spazmları ve hareketlerine neden olabilir. Pek çok insan sonunda
mobilite yardımına ihtiyaç duyar. Bu formu olan bazı insanlarda ilk belirgin
semptom şizofreni gibi ciddi bir psikiyatrik bozukluktur. Zarar görmüş zihin
veya demans gelişebilir veya gelişmeyebilir. Geç başlangıç formuna sahip bazı
kişiler hastalık nedeniyle kısaltılmış bir ömre sahiptir, diğerleri ise
değildir.
Teşhis
Tay-Sachs hastalığının teşhisi, kapsamlı
bir klinik değerlendirme ve vücuttaki heksosaminidaz A seviyelerini ölçen kan
testleri gibi özel testlerle doğrulanabilir. Heksosaminidaz A, Tay-Sachs
hastalığına sahip kişilerde azalır ve çocukluk formunda yoktur veya neredeyse
yoktur. Moleküler genetik testler, Tay-Sachs hastalığının teşhisini doğrulayabilir.
Moleküler genetik testler, hastalığa neden olduğu bilinen HEXA genindeki
mutasyonları tespit edebilir, ancak sadece uzman laboratuvarlarda bir teşhis
servisi olarak kullanılabilir. Bazı durumlarda, amniyosentez ve koryon villus
örneklemesi (CVS) gibi özel testlere dayanarak doğumdan önce (doğum öncesi)
Tay-Sachs hastalığının tanısından şüphelenilebilir. Amniyosentez sırasında,
gelişmekte olan fetüsü çevreleyen bir sıvı numunesi çıkarılırken, CVS
plasentadan bir bölümünden doku numunelerinin çıkarılmasını içerir. Bu
örnekler, hekzosaminidaz A’nın mevcut olup olmadığını veya Tay-Sachs hastalığı
olan kişilerde olduğu gibi, büyük ölçüde azalmış seviyelerde bulunup
bulunmadığını belirlemek için incelenmiştir. Buna enzim deneyi denir. Doğum
öncesi tanı, eğer HEXA geninde spesifik hastalığa neden olan mutasyon ailede
bilinirse, CVS veya amniyosentez yoluyla elde edilen doku örneklerinin
moleküler genetik testleriyle de mümkündür. Kan testleri, bireylerin Tay-Sachs
hastalığı için taşıyıcı olup olmadığını belirleyebilir (yani, hastalık geninin
bir kopyasına sahiptir). Tay-Sachs hastalığı olan bireylerin akrabaları,
hastalık geninin taşıyıcı olup olmadıklarını belirlemek için test edilmelidir.
Bir çocuk sahibi olmayı ve herhangi bir Yahudi soyuna sahip olmayı planlayan
çiftlerin (sadece Aşkenazi değil) hamilelikten önce taşıyıcı taramasından
geçmeleri teşvik edilir.
Tedavi
Tay-Sachs hastalığının spesifik bir
tedavisi yoktur. Tedavi, her bir bireyde belirgin olan spesifik semptomlara
yöneliktir. Tedavi, uzman bir ekibin koordine çabalarını gerektirebilir. Çocuk
doktorları, nörologlar, konuşma patologları, işitme problemlerini gören ve
tedavi eden uzmanlar (odyologlar), göz uzmanları ve diğer sağlık
profesyonellerinin etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir
şekilde planlaması gerekebilir. Genetik danışma, etkilenen bireyler ve aileleri
için faydalı olabilir. Tüm aile için psikososyal destek önerilmektedir.
Beslenme zorluğu potansiyeli nedeniyle, bebeklerin beslenme durumu ve uygun
hidrasyon için izlenmesi gerekir. Beslenme desteği ve takviyesi gerekli
olabilir ve bazen bir besleme tüpünün yerleştirilmesi gerekebilir. Beslenme
desteğine ek olarak, yiyecek, sıvı veya diğer yabancı maddelerin yanlışlıkla
akciğerlere (aspirasyon) girmesini önlemek için bir besleme tüpü gerekebilir.
Antikonvülsanlar, Tay-Sachs hastalığına sahip bazı kişilerde ortaya çıkan
nöbetleri tedavi etmek için kullanılabilir, ancak her insanda etkili
olmayabilir. Ayrıca, nöbetlerin tipi ve sıklığı, bir kişinin ilaç tipinde veya
dozajında değişiklik gerektirecek şekilde değişebilir.
Sıklık
Tay-Sachs hastalığı genel popülasyonda çok
nadir görülür. Bu hastalığa neden olan genetik mutasyonlar Aşkenazi (doğu ve
orta Avrupa) Yahudi mirası insanlarında diğer kökenden daha yaygındır. Bu
hastalıktan sorumlu mutasyonlar, Quebec’in bazı Fransız-Kanadalı
topluluklarında, Pennsylvania’daki Eski Düzenli Amish topluluğunda ve
Louisiana’nın Cajun popülasyonunda da daha yaygındır.
Kalıtım Paterni/Deseni
Bu durum otozomal resesif bir kalıtsal kalıtımla
ifade edilir, yani her bir hücredeki genin her iki kopyası da mutasyonlara
sahiptir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri
mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun
belirtilerini ve semptomlarını göstermezler.
Hastalığın Diğer Adları
GM2 gangliosidosis, tip 1
HexA eksikliği
B varyantı GM2
gangliosidozu
Heksosaminidaz A
eksikliği
Heksosaminidaz alfa-alt
birim eksikliği (değişken B)
Wernicke-Korsakoff Sendromu tiyamin (B vitamini) eksikliğinde oluşan bir zihinsel
hastalıktır. Wernicke-Korsakoff Sendromu hem Wernicke Ansefalopatisi (Wernicke
Sendromu) hem de Korsakoff Sendromuyla ilişkilidir. Wernicke ve Korsakoff
Sendromu arasındaki ilişki hakkında farklı görüşler vardır. Bazı kaynaklara
göre birbirlerinden ayrı olarak gelişen ilişkili sendromlardır; ancak bazı
kaynaklara göre de aynı hastalığın farklı aşamalarıdır. Eğer bu iki sendrom
aynı hastalığın farklı aşamaları olarak değerlendirilirse; Wernicke
Ansefalopatisi akut fazı, Korsakoff Sendromu da kronik fazı temsil etmektedir.
Wernicke Ansefalopatisi üç temel özelliği göstermesi üzerinden
tanımlanmakta olan nörolojik bir hastalıktır. Bu özellikler: kaygı, kas
koordinasyon bozukluğu ve anormal göz hareketleri. Korsakoff Sendromu, daha az hastalarda
gözlemlenmektedir. Korsakoff Sendromu bir çeşit bunamadır. Korsakoff Sendromu, hafıza
kaybı ve konfabülasyon (hafızadaki boşlukları hastanın kendi bilincinde
yarattığı gerçek olmayan anılar ile doldurması) gibi iki temel özellik
üzerinden tanımlanmaktadır.
Wernicke-Korsakoff Sendromu genellikle kronik alkol kullanımının sonucu
olarak ortaya çıkmaktadır. Ancak yetersiz beslenme, uzun süreli kusma, yeme
bozuklukları, kanser, AIDS, enfeksyon gibi sistem hastalıkları, obezite
cerrahisi, doku nakli veya kemoterapi sonucunda da oluşabilmektedir.
Araştırmalara göre Wernicke-Korsakoff Sendromu’nun ortaya çıkmasında genetik
yatkınlık da etkili olabilmektedir.
Belirtileri ve
Semptomları
Tıbbi literartüre göre Wernicke Sendromu’nun bir episodunun Korsakoff
Sedromu’nun ilerlemesini tetikleyip tetiklemediği yönünde bir netlik yoktur. Wernicke
Sendromunun ana semptomlarını oluşturan kafa karışıklığı ve oryantasyon
bozukluğu birkaç gün veya hafta içinde gelişmektedir. Wernicke Sendromuna sahip hastalarda uyuşukluk (letarji),
dikkatsizlik, baş dönmesi ve yön duygusunu yitirmeye rastlanmaktadır. Nadir
olarak dalgınlık hali, düşük tansiyon, hipotermi ve komaya da
rastlanabilmektedir. Wernicke Sendromu hastalarında rastlanan ataksi (kas
koordinasyon bozukluğu) yürüş şeklini de etkilemektedir. Bu etki bazı
hastalarda yavaş ve düzensiz yürüme olarak ortaya çıkmaktadır. Hastalığın akut
devresinde hasta birey desteksiz yürüyemez veya ayakta duramaz hale
gelebilmektedir. Çift görme, göz titremesi (nistagmus), göz kaslarının paralizi
(oftalmopleji) ve bazı nadir vakalarda da üst göz kapağında düşüklük
Wernicke Sendromu’nda görülen göz anomalileridir.
Korsakoff Sendromu, çoğunlukla Wernicke Sendromu’na da sahip alkol bağımlılarında
ortaya çıkmaktadır. Wernicke Sendromu’na sahip insanların yüzde 80-90’ında
Korsakoff Sendromu’na da rastlanmaktadır. Korsakoff Sendromu’na sahip çoğu
hastada Wernicke Sendromu’nda görülen beyin lezyonlarına rastlanmaktadır.
Korsakoff Sendromu’na sahip bireyler hasta olduklarının farkında değillerdir. Korsakoff
Sendromu’nun belirtileri genellikle Wernick Sendromu’nun belirtileri azalmaya
başlıyınca ortaya çıkmaktadır. Korsakoff Sendromu’nun semptomları arasında
hafıza kaybı, yeni anı oluşturamama, unutkanlık, halüsinasyon, oryantasyon
bozukluğu ve görüş bozukluğu bulunmaktadır. Hasta bireylerde nadiren
konfabülasyona (hafızadaki boşlukları hastanın kendi bilincinde yarattığı
gerçek olmayan anılar ile doldurması) da rastlanmaktadır.
Wernicke-Korsakoff Sendromu’nda görülen ek semptomlar arasında birçok
farklı siniri etkileyen hastalıklar (polinöropati) ve merkezi sinir sistemi dışındaki sinirleri etkileyen
hastalıklar (periferik
nöropati) bulunmaktadır. Periferik
nöropati, kollarda ve
bacaklarda güçsüzlüğe ve yürümede güçlüğe neden olabilmektedir. Wernicke-Korsakoff
Sendromu’u görülenlerde çeşitli kardiyovasküler rahatsızlıklara da
rastlanabilmektedir. Bu rahatsızlıklar kalp çarpıntısı (takikardi), uzun süre
ayakta kalmaya bağlı düşük tansiyon ve bilinç kaybıdır (senkop).
Wernicke-Korsakoff Sendromu hastalarında sosyal davranışlar ve dikkat zarar
görmeden korunmaktadır. Hasta bireyler sosyal becerilerine ve konuşma
yetilerine sorunsuzca sahip olmaya devam etmektedirler. Bu yüzden başka
insanlar tarafından hasta olduğu bilinmeyen hasta bireyin hasta olduğu sosyal
becerilerine bakılara anlaşılamamaktadır.
Nedenleri
Wernicke-Korsakoff Sendromu tiyamin (B vitamini) eksikliğinde oluşmaktadır.
Tiyamin, enerji metabolizması için önemli olan bazı enzimlerlele eş değere
sahiptir. Vücudun tiyamin ihtiyacı metabolizma hızına bağlı olmakla beraber
metabolizma ihtiyacının ve glükoz alımının yüksek olduğu durumlarda bu ihtiyaç
artmaktadır. Tiyamin, beyindeki enerji üretiminde önemli bir rol oynamaktadır.
Bu yüzden de eksikliğinde beyindeki metabolizmasal faaliyetlerin, yüksek
metabolizma ihtiyacına ve tiyamin eksilişine bağlı olarak engellenmesiyle nöronal
hastalılar oluşmaktadır.
Alkol tiyaminin vücut tarafından emilmesini ve tiyaminin karaciğerde
depolanmasını azaltmakta olup; tiyamini aktif hale getiren enzimin çalışmasını
engellmektedir. Bu yüzden de alkol bağımlılarında Wernicke-Korsakoff Sendomu’na
rastlanma olasılığı yüksektir.
Hastalık, yanlış beslenme ve yeme bozukluklarından da
kaynaklanabilmektedir. Açlık (starvasyon), yeme
bozukluğu (örn. anoreksi), uzun süren kronik kusma gibi semptomlar
Wernicke-Korsakoff Sendromu’nda görülen yeme bozukluklarındandır. Kanser, AIDS,
gastropati, diyaliz gerektiren böbrek hastalıkları gibi hastalıklar da
Wernicke-Korsakoff Sendromu’na neden olabilmektedir.
Bazı vakalarda kalıtsal faktörler de Wernicke-Korsakoff sendromuna neden
olan genetik yatkınlığın ortaya çıkasını sağlayabilmektedir. Ancak genetik
yatkınlığın hastalığın ortaya çıkmasında bir rolü olup olmadığını anlamak için
daha çok çalışmaya ihtiyaç vardır.
Hastalığın Görülme
Sıklığı
Wernicke-Korsakoff Sendromu erkeklerde kadınlara göre daha çok görülmekte
olup, 30-70 yaş aralığında da daha sık rastlanmaktadır.
Teşhis Konulması
Wernicke-Korsakoff Sendromu’nun tanısı detaylı hasta geçmişi, rutin
laboratuvar testleri, karaciğer fonksiyon testi gibi bu semptomları gösteren
başka hastalıkların değerlendirmeye alınmamasını sağlayacak klinik
değerlendirme ve incelemelerden sonra konmaktadır. Tanının konması için tiyamin
ve alyuvar transketolaz faaliyetlerini/aktivitesini ölçen testler de
yapılmaktadır. Çünkü tiyamin ve alyuvar transketolazı Wernicke-Korsakoff
sendromunun görülmesi durumunda düşüşe uğramaktadır. Bilgisayarlı tomografi
(CT) taraması ve manyetik rezonans görüntülemesi (MRI) semptomların tümör,
infarktüs ve kanama gibi nedenlerden dolayı oluşmadığını kesinleştirmek için
gereklidir. CT taraması ve MRI Wernicke-Korsakoff sendromuna bağlı olarak
beyinde oluşan değişimlerin görüntülenmesini sağlayabilmektedir.
Tedavi
Edilmesi
Tedavinin ana
amacı semptomları olabildiğince kontrol altına almak ve hastalığın ilerleyişini
durdurmaktır. Semptomları kontrol altına almak amacıyla bazı hastaların
başlangıçta hastaneye yatırılması gerekebilmektedir. Wernicke Ansefalopatisi
akut bir sendromdur, ölüm ve nörolojik problemlerin önüne geçilebilmesi için
tedaviye acilen başlanması gerekmektedir.
Wernicke ansefalopatisine
sahip hastalar tiyamin verilişi, doğru beslenme ve sıvı verilişi ile tedavi edilmektedir.
Tiyaminin direkt olarak damardan verilmesi gerekmektedir. Çünkü hastalarda tiyaminin
abdominal emilimi gecikmektedir. Alkol bağımlısı insanlarda az olan magnezyum
ve potasyum gibi elektotlar da tiyamin ile beraber ekstradan verilebilir.Mental
durum değişiklikleri, görüş bozuklukları ve kas koordinasyon bozuklukları
tiyaminin verilmeye başlanması ile geçmeye başlamaktadır. Tiyamin verilişi günlük olarak birkaç ay devam edebilmektedir.
Uzun süreli
alkol kullanımı Wernicke-Korsakoff Sendromu’nun ortaya çıkmasındaki ana
nedenlerden biri olduğu için alkol alımının durdurulması gerekmektedir. Alkol
alımının durdurulmasıyla beraber gerekli beslenme değişikliklerin de yapılması
gerekmektedir. Wernicke ansefalopatisi hastaları tek başlarına hareket etmekte
güçlük çekecekleri için tedavinin ilk safhalarında hareket etmelerinde yardımcı
olacak birilerine ihtiyaç duyabilirler. Ayrıca hastaların hareket etmesini daha
kolay hale getirmek için fizik tedeaviye ihtiyaç duyulabilir. Tedaviye
başlanılan hastalık safhasına göre yürüme güçlükleri kalıcı olabilmektedir. Korsakoff
Sendromu hastaları çok nadiren iyileşme göstermektedir. Çoğu hastanın günlük
aktivitelerini gerçekleştirebilmesi için dışarıdan bir desteğe ihtiyacı vardır.
Referanslar
Wernicke-Korsakoff
syndrome. (n.d.). Retrieved June 27, 2019, from
Anensefali, beyin ve kafatası kemiklerinin
anormal gelişimi ile karakterize bir nöral tüp defektidir. Nöral tüp, normal
bir gebeliğin 3. Ve 4. Haftaları arasında kapanan ve bununla beraber embriyonun
beynini ve omuriliğini oluşturan dar bir geçittir. Nöral tüpün kafayı oluşturan
kısmı kapanamadığında, beynin, kafatasının ve üstünü örten saçlı derinin büyük
bir kısmının yokluğu Anensefali olarak adlandırılır. Bu hastalığa sahip
bebekler, önbeyin ve serebrum olmadan doğarlar. Bu eksiklikler genellikle bu
bebeklerin görme ve işitme engelli olmalarına, bilinçten ve acı hissetme
yetisinden yoksun olmalarına sebep olur. Anensefaliye sahip neredeyse her bebek
anne karnında ölmektedir. Bazı bebekler doğumdan sonra birkaç saat veyahut
birkaç gün yaşayabilmektedir.
Genetik
Değişikler/ Etken Faktörler
Anensefalinin altında yatan nedenler henüz
tam olarak anlaşılamamıştır. Diğer nöral tüp defektleri gibi anensefalinin de
birden fazla genle ve çevresel faktörle alakalı olduğu düşünülmektedir.
MTFHR geni, vücutta Folat (Vitamin B9)
üretimini sağlayan gendir. Bu vitaminin eksikliği nöral tüp defektlerinden
sorumlu bir etkendir. Gebelikten önce ve gebelik süresince folik asit takviyesi
almak nöral tüp defektlerini azaltmakta yardımcı olmaktadır.
Aynı zamanda gebede diyabet varlığı,
obezite varlığı, yüksek sıcaklıklara uzun süre maruz kalma (ateşli
hastalık/sauna/hamam), belli başlı nöbet önleyici ilaçlar nöral tüp defekti
oluşma riski meydana getirirler.
Belirti
ve Semptomlar
Anensefalik kafatası
görünümü
Anensefaliye sahip
neonatlarda ayrıca renal yetmezlik ve spina bfida da sık izlenir.
Genetik
Görülme Sıklığı
Anensefali oldukça sık görülen bir nöral
tüp defektidir. Sıklığı 1000 gebelikte 1’dir.
Kalıtım
Paterni
Anensefalide sporadik vakalar izlenir.
Bunun anlamı hastalığa sahip olan hastalarda aile öyküsüne rastlanılmamasıdır.
Kalıtım paterni belirsizdir.
Teşhis
Yöntemleri/ Tedaviler
Prenatal tanı mümkündür, tedavi
bulunmamaktadır. Hastalığın prognozu oldukça kötü olduğundan gebelikler
genellikle abortus (düşük) ile sonuçlanır.