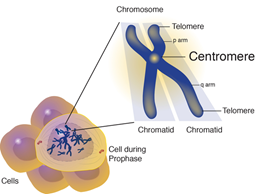
Diğer İsimler
Li ve Fraumeni Sarkoma ailesi sendromu; SBLA sendromu (Sarkom, Meme, Lösemi ve Adrenal Bezi); LFS1; Li Fraumeni sendromu
Kategoriler
Konjenital ve Genetik Hastalıklar; Endokrin Hastalıkları; Kalıtsal Kanser Sendromları; Sinir Sistemi Hastalıkları; Nadir Kanserler
Bu hastalık altında gruplandırılmıştır: Ailesel meme kanseri
Genel Bilgi
Li-Fraumeni sendromu (LFS), çeşitli nadir
görülen kanser türlerine karşı kalıtsal bir ailevi yatkınlıktır. Bu TP53 olarak
bilinen bir tümör baskılayıcı genindeki bir değişiklik (mutasyon) nedeniyledir.
Gen tarafından üretilen ortaya çıkan p53 proteini hasar görmüştür (veya başka
şekilde işlev görmez hale getirilmiştir) ve kötü huylu tümörlerin gelişmesini
önlemeye yardımcı olamamaktadır. Çocuklar ve genç yetişkinler, özellikle
yumuşak doku ve kemik sarkomları, meme kanseri, beyin tümörleri, adrenokortikal
karsinom ve akut lösemi olmak üzere birçok kansere karşı hassastır. LFS
hastalarında görülen diğer kanserler gastrointestinal kanserleri ve akciğer,
böbrek, tiroid ve derinin yanı sıra gonadal organlardaki (yumurtalık, testis ve
prostat) kanserleri içerir.
TP53 gen mutasyonu olan herkesin mutlaka
kanser geliştirmeyeceğini, ancak risklerin genel popülasyonda olduğundan çok
daha yüksek olduğunu not etmek önemlidir. LFS tanısı kritik öneme sahiptir, bu
nedenle etkilenen ailelerin erken kanser teşhisi için tarama yapmanın yanı sıra
uygun genetik danışmanlık alması da mümkündür.
İşaretler
ve Belirtiler
Birinin LFS’de öne çıkan kişisel veya aile
kanser öyküsü varsa, LFS’den şüphelenilebilir. Ek olarak, klinisyenleri LFS
tanısı potansiyeli konusunda uyarması gereken sendromun karakteristik olan bazı
nadir kanserleri vardır. Çoklu çocukluk çağı kanserli hastalar veya aileleri veya
adrenokortikal veya koroid pleksus karsinoması gibi spesifik nadir kanserler,
uygulayıcıları LFS gibi kalıtsal bir kanser sendromu potansiyeli konusunda
uyarmalıdır. Her ne kadar kalıtsal bir kanser sendromu olarak tanımlanmasına
rağmen, tüm doktorlar LFS teşhisinin farkında değildir.
LFS ile en yakından ilişkili kanserler
(çekirdek kanserler):
• Yumuşak doku sarkomu
• Osteosarkom
• Meme kanseri
• Beyin ve CNS tümörleri (glioma, koroid
pleksus karsinomu, SHH alt tipi medulloblastom, nöroblastom)
• Adrenokortikal karsinom
• Akut lösemi
Diğer kanserler de görülebilir, ancak riskler
çekirdek kanserlere göre daha düşüktür:
• Akciğer adenokarsinomu
• Melanom
• Gastrointestinal tümörler (kolon, pankreas
gibi)
• Böbrek
• Tiroid
• Gonadal germ hücreleri (yumurtalık, testis
ve prostat gibi)
LFS’li bireyler, 40 yaşına kadar kansere
yakalanma oranının yaklaşık % 50’sine ve 60 yaşına kadar yüzde 90’a ulaşma ihtimaline
sahipken, kadınlar belirgin bir şekilde artan meme kanseri riskinden dolayı
yaşamları boyunca kanser riski % 100’dür. LFS’li birçok kişi yaşamları boyunca
iki veya daha fazla primer kanser geliştirir.
Nedenleri
CHEK2 ve TP53 genleri, Li-Fraumeni sendromu
ile ilişkilidir.
Li-Fraumeni sendromlu tüm ailelerin yarısından
fazlası TP53 geninde mutasyon geçirmiştir. TP53, bir tümör baskılayıcı gendir,
yani normalde hücrelerin büyümesini ve bölünmesini kontrol etmeye yardımcı
olduğu anlamına gelir. Bu gendeki mutasyonlar, hücrelerin kontrolsüz bir
şekilde bölünmesine ve tümör oluşturmasına izin verebilir. Diğer genetik ve
çevresel faktörlerin de TP53 mutasyonu olan kişilerde kanser riskini etkilemesi
muhtemeldir.
Li-Fraumeni sendromu ve Li-Fraumeni benzeri
sendromu karakteristik kanserli birkaç ailede TP53 mutasyonu yoktur, ancak
CHEK2 geninde mutasyon vardır. TP53 geni gibi, CHEK2 de bir tümör baskılayıcı
gendir. Araştırmacılar CHEK2 mutasyonlarının gerçekte bu koşullara neden olup
olmadığından emin değiller veya yalnızca belirli kanser risklerinin artmasıyla
(meme kanseri dahil) ilişkilendirdiler.
Miras
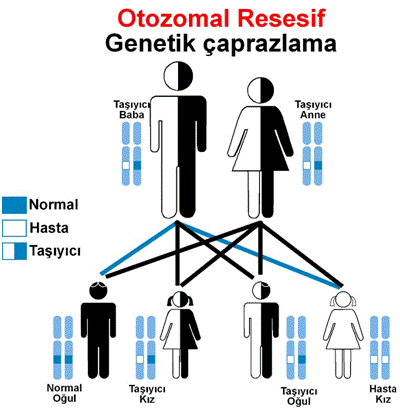
Li-Fraumeni sendromu (LFS), otozomal dominant
bir şekilde kalıtsaldır. Bu, bir kişinin sadece her hücrede sorumlu genin bir
kopyasında LFS’ye sahip olması için bir değişikliğe (mutasyona) ihtiyacı olduğu
anlamına gelir. Bazı durumlarda, etkilenen bir kişi etkilenen bir ebeveynin
mutasyonunu devralır. Diğer durumlar, gendeki yeni (de novo) mutasyonlardan
kaynaklanabilir; bu vakalar ailelerinde düzensizlik öyküsü olmayan kişilerde
görülür. LFS’li bir kişinin, değiştirilen gen boyunca çocuğuna geçmesiyle her
hamilelikte% 50 ihtimal vardır.
Görülme
sıklığı
Li-Fraumeni’nin tam prevalansı
bilinmemektedir. ABD’deki Li-Fraumeni sendromu hastalarının bir kaydı, 64
aileden yaklaşık 400 kişinin bu rahatsızlığa sahip olduğunu göstermektedir.
Teşhis
Genetik veya nadir bir hastalık için tanı
koymak genellikle zor olabilir. Sağlık uzmanları tipik olarak bir tanı koymak
için bir kişinin tıbbi geçmişine, semptomlarına, fizik muayenesine ve
laboratuvar test sonuçlarına bakar. Aşağıdaki kaynaklar bu durum için teşhis ve
test ile ilgili bilgi sağlar. Teşhis konması hakkında sorularınız varsa, bir
sağlık uzmanına başvurmalısınız.
Test Kaynakları
Genetik Test Kayıt Defteri (GTR), bu durum
için genetik testler hakkında bilgi sağlar. GTR için hedef kitle, sağlık
hizmeti sağlayıcıları ve araştırmacılardır. Genetik test hakkında özel soruları
olan hastalar ve tüketiciler bir sağlık kuruluşu veya genetik uzmanı ile
irtibata geçmelidir.
Standart
Terapiler
Tedavi
Şu anda, LFS veya germline TP53 gen mutasyonu
için standart bir tedavi yoktur. Bazı istisnalar dışında, LFS’li kişilerdeki
kanserler, diğer hastalardaki kanserler ile aynı şekilde tedavi edilir, ancak
araştırmacılar LFS’de yer alan kanserleri en iyi nasıl yönetebileceği ile
ilgili araştırmalara devam eder.
Araştırmalar, LFS’li bireylerin radyasyona
bağlı kanserler için yüksek risk olarak göründüğünü, bu nedenle radyoterapinin
kullanımına dikkatle yaklaşılması gerektiğini belirtti. Bu nedenle,
bilgisayarlı tomografi (BT) taramaları ve iyonlaştırıcı radyasyonu içeren diğer
tanı teknikleri sınırlı olmalıdır. Bununla birlikte, faydalar risklerden ağır
basarsa radyasyon terapisinden kaçınılmamalıdır.
LFS ile yaşayanlar, bir dizi farklı kanserin
gelişimine duyarlı olduklarından, bireyler güneşten korunma ve tütün
ürünlerinden kaçınma gibi sağlıklı bir yaşam tarzı için basit önlemler
almalarını sağlamalı.
Erken kanser tespitinin genel sağkalımı büyük
ölçüde artırabileceği ve LFS tanısı konan kişilerin önleyici taramaya uyması
gerektiği yaygın olarak kabul edilmiştir. LFS araştırmacıları, onkologları ve
genetik danışmanlarından oluşan uzman bir panel, Li-Fraumeni sendromunun
tanımına uyan hastalar için tüm vücut MRG taramasını kullanan gözetim önerileri
yayınladı. Bu, LFS teşhisi konulduktan hemen sonra yapılmalıdır. Kısaca, tarama
önerileri şunları içermektedir:
Çocuklar
• Genel değerlendirme
o Her 3-4 ayda bir fiziksel muayeneyi
tamamlayın
o Tıbbi kaygılar için birinci basamak hekimi
ile hızlı değerlendirme
• Adrenokortikal karsinom
o Her 3-4 ayda bir karın ve pelvis ultrasonu
o Yetersiz ultrason durumunda, her 3-4 ayda
bir kan testleri
• Beyin tümörü
o Yıllık beyin MRG (kontrastlı ilk MRG – daha
sonra önceki MRG normal ve yeni bir anormallik yoksa kontrastsız)
• Yumuşak doku ve kemik sarkomu
o Yıllık tüm vücut MRG
Yetişkinler
• Genel değerlendirme
o Her 6 ayda bir fiziksel muayeneyi tamamlayın
o Tıbbi kaygılar için birinci basamak hekimi
ile hızlı değerlendirme
•Meme kanseri
o Meme farkındalığı (18 yaş ve üstü)
o Yılda iki kez klinik meme muayenesi (20 yaş
ve üstü)
o Yıllık meme MRG taraması (20-75 yaş arası) –
ideal olarak, yıllık tüm vücut MRG’si ile dönüşümlü olarak (her 6 ayda bir
tarama)
o Risk azaltıcı bilateral mastektomi düşünün
(Ultrason ve mamografi kullanımının kullanılmadığını unutmayın)
• Beyin tümörü (18 yaş ve üstü)
o Yıllık beyin MRG (kontrastlı ilk MRG – daha
önceki MRG normal ise kontrastsız)
• Yumuşak doku ve kemik sarkomu (18 yaş ve
üstü)
o Yıllık tüm vücut MRG
o Her 12 ayda bir karın ve pelvis ultrasonu
• Gastrointestinal kanser (25 yaş ve üstü)
o 2-5 yılda bir üst endoskopi ve kolonoskopi)
• Melanom (18 yaş ve üstü)
o Yıllık dermatolojik muayene
Ayrıca, meme kanserinin 20 yaşında veya
civarında ortaya çıkmış olduğu aileler için farkındalık ve tarama bilinen en
erken başlangıç yaşından 5 ila 10 yıl önce düşünülebilir. Aynısı
gastrointestinal kanserler için de önerilmektedir. Ailede gastrointestinal
kanserin bilinen en erken başlangıcından 5 yıl önce taramayı düşünün.
Araştırma
Terapileri
İşlevsel olmayan TP53 proteinini yeniden
etkinleştirmek veya değiştirmek için küçük moleküllü ilaçları kullanan çok
sayıda strateji aktif olarak çalışılmaktadır, ancak henüz LFS hastaları ile
yapılan klinik çalışmalarda araştırılmamıştır.
Bir
Uzman Bulun
Tıbbi yardıma ihtiyacınız olursa, bu
hastalıkta deneyimi olan doktorları veya diğer sağlık profesyonellerini
arayabilirsiniz. Bu uzmanları savunma kuruluşları, klinik deneyler veya tıbbi
dergilerde yayınlanan makaleler aracılığıyla bulabilirsiniz. Ayrıca
bölgenizdeki bir üniversite veya üçüncül tıp merkezi ile iletişim kurmak
isteyebilirsiniz, çünkü bu merkezler daha karmaşık vakalar görmeye ve en son
teknoloji ve tedavilere sahip olma eğilimindedir.
Bölgenizde bir uzman bulamıyorsanız, ulusal
veya uluslararası uzmanlarla iletişim kurmayı deneyin. Sizi, bildiği birisine
konferanslar veya araştırma çalışmaları yoluyla yönlendirebilirler. Bazı uzmanlar,
sizinle ilgilenmek için seyahat edemiyorsanız, telefonla veya e-posta yoluyla
sizinle veya yerel doktorlarınızla görüşmek isteyebilir.
Sağlık
Kaynakları
Genetik konusunda uzmanlaşmış bir tıp uzmanı
bulmak için, doktorunuza sevk isteyebilir veya kendiniz için arama
yapabilirsiniz. Çevrimiçi dizinler Amerikan Tıbbi Genetik Koleji ve Ulusal
Genetik Danışmanlar Birliği tarafından sağlanmaktadır. Daha fazla yardıma
ihtiyacınız olursa, bir GARD Bilgi Uzmanı ile iletişime geçin. Ayrıca Genetik
Danışma’dan Genetik danışmalar hakkında daha fazla bilgi edinebilirsiniz.
Kaynaklar