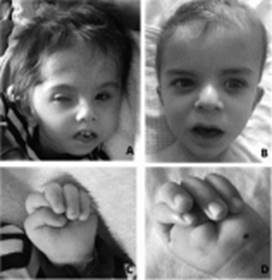
Genel Bilgi
Dandy-Walker malformasyonu beyinciğin ve 4. ventrikülün embriyonik gelişiminde oluşan bir beyin anomalisidir. Beyincik; beyinin, hareketi koordine eden ve idrak ve davranışla ilgilenen bir bölümüdür. Dandy-Walker malformasyonu, “cerebellar vermis” olarak bilinen beyinciğin orta lobunun gelişimde geri kalması (küçücük boyut ve anormal pozisyon), 4. ventrikülde kist büyümesi ve “posterior fossa” olarak bilinen arka çukurun büyümesi ile karakterize edilir. DWM %20-80 oranında omurilik sıvısının normal akışının engellenmesiyle çok miktarda sıvının beyin çevresinde birikmesiyle ortaya çıkan hidrosefali ile ilişkilendirilir. Bu durum kafatasında yüksek basınca ve kafanın şişmesine yol açar ve ayrıca nörolojik bozulmaya da yol açabilir. [1] Bu malformasyona sahip bireylerde motor gelişiminde gecikme, hipotoni ve kas koordinasyon bozukluğu gibi motor eksiklikleri, yarısına yakınında da mental gerilik ve bazılarında da hidrosefali görülür. DWM heterojen bir hastalıktır. Sendromik olmayan DWM için yaklaşık olarak %1-2 deneysel tekrar etme riski olması Mendel genetiğiyle kalıtılma olasılığının düşük olduğunu gösterir. [3]
Genetik Değişiklikler/Etken Faktörler
DWM beyinciğin ve çevresindeki yapıların erken embriyonik dönemdeki gelişiminde bozukluklar olması sonucunda ortaya çıkar. Birkaç hastada 3q24.3 kromozom lokasyonunda (ZIC1 ve ZIC4 olarak bilinen ilk DWM genlerinin lokasyonunda), 6p25 ya da 13q32.2-q33.2 kromozom lokasyonlarında delesyon olması, 9p kromozom lokasyonunda duplikasyon olması gibi kromozom anormallikleri olduğu görülmüştür. Geri kalan kısmında da kardeşlerde tekrar etme sıklığı %5’ten az olması nedeniyle muhtemelen daha kompleks genetik ve çevresel faktörler (teratojenler) sonucu ortaya çıkmaktadır. Birkaç izolasyonlu DWM’ye sahip kardeşler örneği rapor edilmiştir ve bu durumda otozomal çekinik ya da X kromozomuna bağlı kalıtım olduğu orataya sürülmüştür. Fakat bu vakaların çoğunun muhtemelen Serebellar Vermis Hipoplazisi (CVH) olduğu ya da tipik bir DWM olmadığı düşünülmektedir. Bu ailelerde tekrar etme riski %25’e çıkabilecek kadar yüksektir. DWM, birçok doğumdan gelen bozukluğa sahip genetik sendromların bir parçası olarak da ortaya çıkabilir. Bu sendroma örnek olarak yüz hemanjiyomu (kan damarlarında urlar), kalp ve göğüs kemiği bozuklukları ve DWM içeren PHACES sendromu verilebilir. DWM’li daha birçok sendrom ve kromozom anormallikler rapor edilmiştir fakat bunların çoğunda DWM’den ise CVH olduğu görülmüştür. [1]
Araştırmacılar birkaç gende Dandy-Walker malformasyonuna yol açtığı düşünülen mutasyonlar bulmuştur fakat mutasyonlar sadece çok az sayıdaki vakaların sebebidir. Dandy-Walker malformasyonu birçok kromozom anormallikleriyle de ilişkilendirilmektedir. Bu durum, bir hücrede bir kromozomun ekstra kopyasının olması (trizomi) gibi bir durumun sadece bir özelliği olarak da ortaya çıkabilir. Dandy-Walker malformasyonu trizomi 18’e (kromozom 18’in ekstra bir kopyasına sahip olmak) sahip insanlarda sıklıkla görülür. Trizomi 13, trizomi 21 ve trizomi 9’a sahip insanlarda da ortaya çıkabilir. Bu durum, belirli kromozomların bölümlerinin delesyonu ve duplikasyonu ile de ilişkilendirilebilir. Dandy-Walker malformasyonu, belirli genlerdeki mutasyonlardan kaynaklanan genetik sendromların bir özelliği olarak ortaya çıkabilir. Fakat Dandy-Walker malformasyonu ile ilişkilendirilen beyin malformasyonları (bozuklukları) genellikle izole (diğer sağlık problemleriyle ilişkili olmayan şekilde) özellikle ortaya çıkar ve bu vakalarda malformasyonun nedeni genellikle bilinmemektedir. Araştırmacılar Dandy-Walker malformasyonunun doğumdan önce erken gelişimi etkileyen çevresel faktörler yüzünden de ortaya çıkabileceğini öne sürmektedirler. Örnek olarak, fetüsün doğum bozukluklarına (teratojenlere) sebep olabilecek maddelere maruz kalması bu durumun ortaya çıkmasına sebep olabilir. Ek olarak, diyabetli bir annenin sağlıklı bir anneye göre Dandy-Walker malformasyonuna sahip bir çocuğa sahip olma olasılığı daha yüksektir. [2]
Belirti ve Semptomlar
Dandy-Walker malformasyonunun semptomları tipik olarak gelişimsel gecikmeyi, hipotoniyi (gevşek bebek; kaslarda güçsüzlük), ya da spastikliği, düşük koordinasyon ve dengeyi (ataksi), ve bazen geniş baş çevresi ve hidrosefali yüzünden artan kafatası basıklığını içerir. Hastaların yarısından azında mental gerilik görülür, çoğunda şiddetli hidrosefali, kromozom anormallikleri ve diğer doğumdan gelen bozukluklar görülür. Hastaların %15-30’u nöbet geçirirler. Beyindeki solunumun kontrol edildiği merkezlerde bu malformasyon sebebiyle oluşan bir hasar var ise bu solunumda soruna yol açar. Bu durumun şiddetli hidrosefaliye sahip hastalarda genellikle görülme olasılığı yüksektir. Teşhis konulma yaşı hastalığın başlangıç zamanına ve hidrosefalinin şiddetine ve başka doğumdan gelen bozukluklara ya da tıbbi problemlere göre değişir. DWM’nin klinik gösterimler ve resimlemeleri, izole Serebellar Vermis Hipopilazisi (CVH) ve Mega Sisterna Magna (MCM) ile örtüşmektedir ve Dandy-Walker kompleksi olarak bilinen anomaliler spektrumu olarak birlikte sınıflandırılmaktadırlar. Fakat deneyimler bu basitleştirmenin sonuçlar ve genetik riskler hakkında yanlış bilgilendirmeye yol açacağını ortaya çıkarmıştır. CVH, tipik DWM’yi karakterize eden beyincik orta lobunun göze çarpan yukarı doğru bir dönüşü olmadan küçük bir beyincik orta lobunu, 4. ventrikülün sistik genişlemesi ya da posterior fossa’nın genişlemesini içerir. Bu malfarmosyon aynı zamanda potansiyel kafa karıştırıcı bir terim olan Dandy-Wlaker variant olarak da adlandırılır. Sonuçlara ilişkin ulaşılabilir data kısıtlıdır ama bu genellikle tipik DWM’den daha şiddetlidir. MCM beyinciğin normal ya da normale yakın boyutuna rağmen posterior fossa’nın genişlemesi ile karakterize edilir. Bu boyutun büyüklüğü, sıvıların beyinciğin altında ve sıklıkla arkasında birikmesi ile ilişkilendirilir. Bu, normal bir variant olarak rapor edilmektedir fakat deneyimler, DWM ya da CVH’de görülenden genellikle daha az şiddetli olmasına rağmen gelişimsel özürler ile ilişkilendirildiğini ortaya çıkarmıştır. Bu kadar benzeyen bu 3 durumun öngörüsündeki belirsizliğin sebebi doğal çeşitlilik ve bu 3 durumu birbirinden ayırmada yaşanan zorluktur.[1]
Genetik Görülme Sıklığı
Dandy-Wlaker malformasyonunun Birleşmiş Milletler’de görülme sıklığı 25.000-30.000 canlı doğumda 1’dir ve kadınlarda erkeklerden daha çok görülür. [1]
Dandy-Walker malformasyonunun 10.000-30.000 yeni doğandan 1’ini etkilediği tahmin edilmektedir. [2]
Kalıtım Paterni/Deseni
Dandy-Walker malformasyonunun çoğu vakaları sporadiktir, yani ailelerinde hastalık geçmişi olmayan insanlarda meydana gelir. Vakaların küçük bir yüzdeliğinin ailelerde tekrar ettiği görülmüştür fakat Dandy-Walker malformasyonu açık bir kalıtım paternine sahip değildir . Birçok genetik ve çevresel faktörler, hastalığın oluşması riskinin belirlenmesinde rol oynar. Dandy-Walker malformasyonuna sahip kişilerin 1. dereceden akrabalarının (kardeşler ve çocuklar gibi) bu hastalığa sahip olma olasılığı popülasyonun genelinden daha fazladır. Dandy-Walker malformasyonu bir genetik durumun özelliği olarak ortaya çıktığında, bu malformasyon o genetik durumun paterninde kalıtılır. [2]
Teşhis Yöntemleri ve Tedaviler
Dandy Walker malformasyonu ultrason, bilgisayarlı tomografi ve MRI kullanırak teşhis edilir. Dandy Walker malformasyonunun doğum öncesi teşhisi de bazen ultrason ve fetal MRI kullanılarak konulur.
Dandy Walker malformasyonu ile ilişkili hidrosefali beyni saran sıvının ameliyatla bir tüp yerleştirilerek yeniden yönlendirilmesiyle ve çekilen sıvının o sıvıyı absorbe edebilecek başka vücut bölümlerine yönlendirilmesiyle tedavi edilir. Dandy Walker malformasyonuna sahip çocuklara, destekleyici bir takım yaklaşımı garanti edilir ve bu yaklaşım özel bir eğitimi, fiziksel terapiyi ve diğer tıbbi, sosyal ya da mesleki servisleri içerir.
Dandy Walker malformasyonuna sahip çocuğu olan ailelere genetik danışmanlık almaları önerilir. [1]
Hastalıkla İlişkili Genler
Birkaç hastada 3. kromozomun kuyruk kısmının 24.3 lokasyonunda yani ZIC1 ve ZIC4 olarak bilinen genlerde delesyon olması Dandy-Walker malformasyonu ile ilişkilendirilmiştir. [1]
Hastalığın Diğer İsimleri
- Dandy-Walker Kisti
- Dandy-Walker Deformasyonu
- Dandy-Walker Sendromu
- DWM
- Hidrosefali, İç, Dandy-Walker Tip
- Hidrosefali, Nonkomünikatif, Dandy-Walker Tip
- Luschka-Magendie Foramen Atrezisi [1]
- Dandy-Walker Kompleksi
- DWS [2]
Kaynaklar
[1] “Dandy Walker Malformation.” NORD (National Organization for Rare Disorders), rarediseases.org/rare-diseases/dandy-walker-malformation/. Erişim Tarihi:25 Eyl. 2019
[2] “Dandy-Walker Malformation – Genetics Home Reference – NIH.” U.S. National Library of Medicine, National Institutes of Health, ghr.nlm.nih.gov/condition/dandy-walker-malformation#synonyms. Erişim Tarihi:25 Eyl. 2019
[3] Murray, J. C., et al. “Dandy-Walker Syndrome-DWS.” OMIM, 1985, www.omim.org/entry/220200?search=Dandy%2BWalker%2BMalformation&highlight=dandy%2Bmalformation%2Bwalker. Erişim Tarihi:25 Eyl. 2019