Genel Bilgi
Balo Hastalığı Multipl Skleroz’un (MS) nadir görülen ve ilerleyen bir çeşididir. Genellikle yetişkinlikte görülür, ancak çocukluk vakaları da bildirilmiştir. Multipl skleroz tipik olarak bal mumlu ve zayıflayan bir hastalık olmasına rağmen, Balo Hastalığı hızlı ilerici olma eğiliminde olduğundan farklıdır. Belirtileri baş ağrısı, nöbetler, kademeli felç, istemsiz kas spazmları ve bilişsel kaybı içerebilir. Balo hastalığı, beyindeki sinir liflerinin etrafındaki yağ örtüsünün kaybı ve adrenal bezin ilerleyici dejenerasyonu ile karakterize nadir görülen kalıtsal bir metabolik hastalıktır.
Belirti ve Semptomlar
Semptomlar, beynin etkilenen bölgelerine göre değişir. Belirtiler birkaç hafta içinde hızlı bir şekilde veya iki ila üç yıl boyunca daha yavaş ilerleyebilir. Bu bozukluğun semptomları arasında genelleştirilmiş kas güçsüzlüğü (hipotoni), hareketi koordine etme yeteneğinin (ataksi), spastik parsiyel felç ve/veya kollarda veya bacaklarda karıncalanma veya yanma hisleri abartılı refleks yanıtları (hiperrefleksi) olabilir. Çoğu vaka, kas spazmları ve felç dahil olmak üzere, daha sık görülen MS tipinde bulunabilecek semptomların kademeli başlangıcı ile karakterizedir. Etkilenen ve zihinsel bozulma yada fizyolojik anormallikleri içerebilen beyin bölgelerine bağlı olarak diğer nörolojik semptomlar gelişir. Bununla birlikte, en ciddi haliyle Balo Hastalığı, yüksek ateş ve ağrılı baş ağrılarıyla başlayan bulaşıcı bir hastalığın varlığını da önerebilir.
Kaynak: https://images.app.goo.gl/3pSwAkD4iM25F9x88
Genetşk Görülme Sıklığı
Balo’nun hastalığı Asya’da, özellikle Çin ve Filipinler’de yaşayan insanlar arasında en yaygın olanıdır. Yetişkinlerin çocuklardan daha fazla kazanması olasıdır ve hem erkekleri hem de kadınları etkileyebilir. İnsanlar genellikle 30’lu yaşlarda hastalığı yakalarlar.
Kalıtım Deseni
Doktorlar, Balo hastalığını neyin tetiklediğinden emin değil. Vücudunuzun yanlışlıkla sağlıklı dokulara saldırıp şişmesine veya iltihaplanmasına neden olan bir tür otoimmün durum olduğunu düşünüyorlar. Balo hastalığına yakalanan bazı kişiler, diğer semptomları fark etmeden hemen önce yüksek ateşli ve şiddetli baş ağrıları olan bir hastalığa sahiptir. Bu nedenle, doktorlar kesin olarak bilinmese de, bunun bir enfeksiyonla bağlantılı olabileceğini düşünüyorlar.
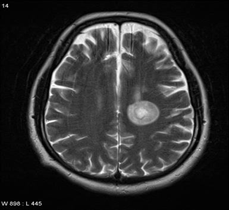
Şekil: Balo disease olan bir hastanın beyninde oluşan lezyonlar (Kaynak: https://images.app.goo.gl/z3heVnSpR1z1qYZF7 )
Teşhis Yöntemleri ve Tedavileri
Balo hastalığı çok nadir olduğu için, beyin ve sinir sistemi (nörolog) problemlerinde uzmanlaşmış bir doktora görünmek en iyisidir. Tıbbi geçmişini ve semptomlarını incelemek gerekiyor. Ayrıca ne kadar iyi hareket ettiğinizi ve kasların bazılarının diğerlerinden daha zayıf olup olmadığını görmek için fiziksel bir muayene edilecektir ve hafızanızın ne kadar iyi olduğunu ne kadar iyi konuşulduğunu kontrol edilecek. Muhtemelen, lezyonları kontrol etmek için beyninizin ve omuriliğinizin manyetik bir rezonans görüntüleme (MRG) taramasını da önerilecek. Vücudunuzun iç kısmının ayrıntılı resimlerini yapmak için güçlü mıknatıslar ve radyo dalgaları kullanır. Ayrıca bir enfeksiyon olup olmadığını kontrol etmek için kan testleriniz olabilir veya doktorun test için belden küçük miktarda spinal sıvı alınabilir. Bazı durumlarda, doktorun uyarılmış bir potansiyel (EP) testi önerebilir.
Hastalığın Diğer İsimleri
Balo Hastalığı
Concentric Sclerosis
Encephalitis Periaxialis
Concentrica Leukoencephalitis
Periaxialis Concentric
Adrenolökodistrofi
Kaynakça
https://rarediseases.org/rare-diseases/balo-disease/
https://www.webmd.com/multiple-sclerosis/balos-disease
https://pdfs.semanticscholar.org/ce23/63690425e3e9d502237cb393bddb54d1cf60.pdf




{kind=link}
{kind=link}
{kind=link}
{kind=link}