Genel bilgi
Glikojen depo hastalıkları, enerji sağlamak ve kandaki glikoz değerini korumak için depolanan glikojenin glikoza dönüştürülemediği bir hastalık grubudur. Genellikle glikojenin glikoza yıkımını sağlayan enzimlerden birinin eksikliğinden ortaya çıkar. Glikojen depo hastalığı tip 1 (GDH 1), sıklığı 1/100 000 olan kalıtsal otozomal çekinik bir hastalıktır. GDH 1 belirtileri ve genetik sebeplerinden dolayı ikiye ayrılır: Glikojen depo hastalığı tip 1A ve glikojen depo hastalığı tip 1B.
Glikojen depo hastalığı tip 1A’da,G6PC genindeki mutasyonlar glikoz-6-fosfataz (G6Paz) enziminde bir eksikliğe neden olur.Endoplazmik retikulum lümeninde glukoz 6 fosfat’ın (G6P) glikoz ve fosfata hidrolizini sağlayan glukoz 6 fosfotaz (G6Paz) enziminin eksikliği glikojen yıkımını engeller. Bu enzim eksikliği glikoz hidrolizini engeller, aynı zamanda vücuttaki diğer önemli metabolitlerin düzenlenmesi ile sonuçlanarak bu lipitlerin, özellikle lipidler ve trigliseritler gibi yağların dengesizliğine veya aşırı birikmesine neden olur. GDH 1A karaciğerde, böbreklerde ve ince bağırsakta glikojen ve yağ birikimine ve bu organlarda işlev bozukluklarına sebep olur.
Belirti ve Semptomlar
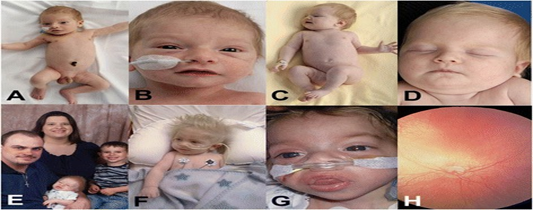
GDH 1A genellikle 3 aylık ve ya 4 yaşları arasındaki bebeklerde gece boyu uyuma ve yenidoğanlarda yeterince beslenmeme sorunlarıyla ortaya çıkar. Etkilenen bebeklerde düşük kan şekeri (hipoglisemi) olabilir ve bu da nöbetlere yol açabilir. Ayrıca vücutta laktik asit birikimi (laktik asidoz), ürik asit (hiperürisemi) adı verilen bir atık ürünün kanda yüksek seviyeleri ve kanda aşırı miktarda yağ (hiperlipidemi) olabilir. Yaşları ilerleyen çocuklarda ince kol ve bacak, kısa boy, gövdesel tipte şişmanlık, ergenliğe geç giriş, karaciğer ve böbrek büyümesi, ishal, deride kolesterol birikimi, taş bebek yüzü, kas zayıflığı, karaciğer büyümesinden dolayı karın şişkinliği gibi etkiler görülebilir.
Genetik Görülme Sıklığı
GDH 1A otozomal resesif bir hastalık olduğu için aynı anormal genin her iki ebeveynden de miras kalması gerekir. Eğer birey bir normal alel gene ve bir de hasta alel gene sahip olursa, bu birey taşıyıcı olur ve genelde semptomları göstermez. Taşıyıcı bireyler hastalığın belirtilerini göstermese de çocuklarına bu geni aktarma riskleri vardır. İki taşıyıcı bireyin çocuklarında bu hastalığın görülme riski %25’dir, çocukların taşıyıcı olma riski ise %50’dir. Otozomal bir hastalık olduğu için kadın ve erkeklerde görülme riski eşittir. Yakın akraba olan ebeveynlerde akraba olmayan ebeveynlere göre daha fazla hasta geni taşıma olasılığı vardır.
GDH hastalığının görülme sıklığı 100 000’de 1’dir. GDH 1A ise GDH 1B’den daha yaygındır ve hastaların %80’ini oluşturur.
Teşhis Yöntemleri ve Tedaviler
Sağlık uzmanları tipik olarak bir tanı koymak için bir kişinin tıbbi geçmişine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına bakar. GSD tip I, anormal glikoz, laktat, ürik asit, trigliserit ve kolesterol seviyelerini gösteren laboratuvar testleri ile teşhis edilir. G6PC geni için moleküler genetik testler tanıyı doğrulamak için kullanılabilir. Moleküler genetik testler ayrıca taşıyıcı testler ve prenatal tanı için kullanılabilir. Moleküler çalışmalar ile erken ve güvenilir tanı olanağı sağlanmakta, taşıyıcılar belirlenebilmekte ve genetik danışma verilebilmektedir. Karaciğer biyopsisi GDH 1A için spesifik enzim eksikliğini kanıtlamak için de kullanılabilir. Karaciğer biyopsi örneklerinde artmış glikojen miktarı ve azalmış G6Paz aktivitesi ve/veya G6Paz geninde mutasyon analizi yapılabilir.
GDH 1A normal glikoz seviyelerini korumak, hipoglisemiyi önlemek ve büyüme ve gelişmeyi en üst düzeye çıkarmak için özel bir diyetle tedavi edilir. Gün boyunca sık sık az porsiyon karbonhidratlar alınmalı ve yaşam boyunca devam etmelidir. Pişmemiş mısır nişastasının beslenmesi kandaki glikoz seviyelerini iyileştirmek için kullanılır. Kandaki ürik asit seviyesini azaltabilen bir ilaç olan allopurinol, ergenlik yıllarında gut benzeri artrit semptomlarını kontrol etmek için yararlı olabilir.
Diyet planı dengeli yapılmalıdır. WHO’nun yaş ve cins için belirlediği ölçütler esas alınmalı ve esansiyel gıdalar mutlaka verilmelidir. Özellikle süt alımı kısıtlandığından kalsiyum ve vitamin D eksikliği yönünden dikkatli olunmalıdır. Ayrıca artmış karbonhidrat metabolizması için yeterli vitamin B1 desteği de yapılmalıdır.
GDH 1A hastaları özel olarak kullanılan böbrek ve karaciğer ultrasonu ve rutin kan çalışmaları ile en az yılda bir kez izlenmelidir.
Genetik danışmanlık, etkilenen bireyler ve aileleri için önerilmektedir.
Erken tanı ve diyet tedavisi ile yeterli metabolik kontrolün sağlanması hem komplikasyonların gelişmesini önleyebilmekte hem de hastaların yaşam kalitesini artırmaktadır.
Hastalıkla İlişkili Gen
G6PC geni (17q21.31)
Hastalığın Diğer İsimleri
GDH 1A
GDH tip 1A
G6Paz eksikliği
Hepatorenal glikojenoz 1A
Glikojen depo hastalığının hepatorenal formu 1A
von Gierke hastalığı 1A
Kaynaklar
- https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/
- https://ghr.nlm.nih.gov/condition/glycogen-storage-disease-type-i#
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=11284&Disease_Disease_Search_diseaseGroup=Glycogen-storage-disease-type-1A&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Glycogen-storage-disease-due-to-glucose-6-phosphatase-deficiency-type-Ia&title=Glycogen%20storage%20disease%20due%20to%20glucose-6-phosphatase%20deficiency%20type%20Ia&search=Disease_Search_Simple
- https://rarediseases.info.nih.gov/diseases/7864/glycogen-storage-disease-type-1a
- https://www.omim.org/entry/232200?search=Glycogen%20storage%20disease%20type%201A&highlight=%28glycogen%7Cglycogenic%29