Sensorinöral sağırlığı olan progresif bulber palsi
Riboflavin taşıyıcı eksikliği
Genel Tanı-Özet
Riboflavin taşıyıcı eksikliği
nöronopatisi, sinir hücrelerini (nöronlar) etkileyen bir hastalıktır. Etkilenen
bireyler tipik olarak, iç kulaktaki sinir hasarından (duyusal sinirsel işitme
kaybı) ve diğer sinirlere verilen hasar belirtilerinden kaynaklanan işitme
kaybına sahiptir. Riboflavin
taşıyıcı eksikliğine, SLC52A2 veya SLC52A3 genlerindeki mutasyonlar neden
olabilir. Kalıtım otozomal resesiftir.Riboflavin tedavisi ile tedavi 2010’dan
beri kullanılmaktadır ve etkili ve muhtemelen hayat kurtarıcı gibi
görünmektedir. Tedavi olmadan, etkilenen bebekler tipik olarak bir yıldan daha
az hayatta kalır.
İç kulaktaki sinirlere ek olarak,
riboflavin taşıyıcı eksikliği nöronopatisi, beynin omuriliğe (beyin sapı) bağlı
olan ve özellikle pontobulbar bölge olarak bilinen bir beyin bölgesinde bulunan
sinir kısmında bulunur. Bu
sinirlere zarar verilmesi, pontobulbar palsi adı verilen bir durum olan,
kendileri tarafından kontrol edilen kasların felç olmasına neden olur.
Pontobulbar bölgedeki sinirler, nefes alma, konuşma ve uzuvları hareket ettirme
dahil olmak üzere birçok gönüllü kas aktivitesini kontrol etmeye yardımcı olur.
Pontobulbar palsinin bir sonucu olarak, riboflavin taşıyıcı eksikliği olan
nöronopatili kişilerde solunum problemleri olabilir; konuşma bozukluğu; ve yüz,
boyun, omuz ve uzuvlardaki kas güçsüzlüğü. Etkilenen bireyler ayrıca kas
sertliği (spastisite) ve abartılı reflekslere sahip olabilir.
Riboflavin taşıyıcı eksikliği
nöronopatisinin başladığı yaş bebeklikten genç erişkinliğe kadar değişmektedir.
Durum bebeklik döneminde başladığında, ilk belirti genellikle hayati tehlike
yaratabilecek sinir hasarının neden olduğu solunum problemleridir. Durum
çocuklarda veya genç erişkinlerde başladığında, ilk önce sensorinöral işitme
kaybı ve ardından pontobulbar palsi bulguları görülür.
Tedavi edilmezse, riboflavin taşıyıcı
eksikliğinin nöronopatisinin belirti ve semptomları zamanla kötüleşir. Şiddetli
solunum problemleri ve solunum yolu enfeksiyonları, bu rahatsızlığı olan
kişilerde normal ölüm nedenidir. Tedavi olmadan, etkilenen bebekler tipik
olarak bir yıldan az yaşarlar. Bununla birlikte, durumu 4 yaşından sonra
geliştirenler genellikle 10 yıldan fazla yaşarlar.
Görülme Sıklığı
Riboflavin taşıyıcı eksikliği nöronopatisi
oldukça nadir görülen bir durumdur. Bilimsel literatürde yaklaşık 100 vaka
bildirilmiştir.
Oluşumunda Etkili Olduğu Düşünülen Temel Etken
Riboflavin taşıyıcı eksikliği
nöronopatisi, SLC52A2 veya SLC52A3 genindeki mutasyonlardan kaynaklanır. Bu
genler, riboflavin taşıyıcıları adı verilen ilgili proteinlerin yapılması için
talimatlar sağlar: RFVT2, SLC52A2 geninden üretilir ve RFVT3, SLC52A3 geninden
üretilir. Her iki protein de, hücre zarı boyunca riboflavin (B2 vitamini olarak
da bilinir) olarak adlandırılan bir vitamini taşır (taşır). Riboflavin vücut
tarafından üretilemez, bu yüzden bir kişinin yediği yiyeceklerden elde
edilmelidir. RFVT3 proteini, ince bağırsak hücrelerinde özellikle yüksek
seviyelerde bulunur ve sindirim sırasında riboflavin emilimi için önemlidir,
böylece vitamin vücutta kullanılabilir. RFVT2 proteini, beyin ve omurilik
hücrelerinde bulunur ve bu dokuların düzgün çalışması için yeterli riboflavin
olmasını sağlamak için önemlidir.
Vücudun hücrelerinde, riboflavin, flavin
adenin dinükleotidi (FAD) ve flavin mononükleotidi (FMN) olarak adlandırılan
moleküllerin çekirdek bileşenidir. FAD ve FMN birçok farklı kimyasal
reaksiyonda yer alır ve çeşitli hücresel işlemler için gereklidir. Bu
moleküllerin önemli bir rolü hücreler için enerji üretimidir. FAD ve FMN,
karbonhidratların, yağların ve proteinlerin parçalanmasında (metabolizması) da
rol oynarlar.
SLC52A2 veya SLC52A3 genindeki
mutasyonlar, riboflavin taşıma kabiliyeti zayıflamış anormal bir riboflavin
taşıyıcı proteine yol açar. Sonuç olarak, vücutta mevcut riboflavin azalması
söz konusudur. Bununla birlikte, bu değişikliklerin riboflavin taşıyıcı
eksikliği nöronopatisinin karakteristik sinir problemlerine nasıl yol açtığı
açık değildir.
Genetik Miras
Riboflavin taşıyıcı eksikliği
nöronopatisi, genellikle otozomal resesif kalıtım paternini izler; bu, her
hücrede genin her iki kopyasında da mutasyon olduğu anlamına gelir. Otozomal
resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış
genin bir kopyasını taşır, ancak bunlar genellikle durumun belirtilerini ve
semptomlarını göstermezler.
Riboflavin tedavisi ile tedavi (yüksek
dozda riboflavin takviyesi) 2010’dan beri kullanılmaktadır ve etkili ve
muhtemelen hayat kurtarıcı gibi görünmektedir. Bu nedenle tedaviye riboflavin
taşıyıcı eksikliğinden şüphelenildiği anda başlamalı ve tanı genetik testlerle
konulmadıkça devam etmelidir. Semptomların iyileşmesi tedaviye başladıktan
sonraki günler ile aylar arasında ortaya çıkabilir ve tedaviye erken başlamanın
daha iyi ve daha hızlı bir iyileşme ile ilişkili olduğu görülmektedir.
Diğer İsimler: Radyal ve patellar aplazi; Radyal ve patellar hipoplazisi; Alınmamış baş parmakları, çıkık eklemler, dar palpebral fissürlerle uzun yüz, uzun ince burun, kemerli damak
Kategoriler: Konjenital ve Genetik Hastalıklar; Kulak Burun Boğaz Hastalıkları; Ağız Hastalıkları; Kas İskelet Hastalıkları
Tanım
Rapadilino sendromu, vücudun birçok bölümünü içeren nadir görülen bir durumdur. Kemik gelişimi özellikle bu durumdan etkilenir ve bu da durumun karakteristik özelliklerinin çoğuna neden olur.
Etkilenen bireylerin çoğu, önkollarda ve
radyal ışın malformasyonları olarak bilinen başparmaklarda kemiklerin az
gelişmiş veya yokluğuna sahiptir. Diz kapağı (patella) da az gelişmiş olabilir
veya bulunmayabilir. Diğer özellikler arasında ağzın çatısında bir açıklık
(yarık damak) veya yüksek kemerli bir damakta; uzun, ince bir burun; ve çıkık
eklemler vardır.
Rapadilino sendromu olan birçok bebek beslenme konusunda güçlük çeker , ishal ve kusma yaşar. Bozulmuş kemik gelişimi ve beslenme problemlerinin kombinasyonu, etkilenen bireylerde yavaş büyümeye ve kısa boylanmaya neden olur.
Rapadilino sendromu olan bazı kişiler, cafe au lait lekeleri olarak bilinen bir deri bulgusuna benzeyen, zararsız açık kahverengi cilt lekelerine sahiptir. Ek olarak, Rapadilino sendromu olan kişilerde, osteosarkom veya lenfoma adı verilen kana bağlı bir kanser türü olarak bilinen bir tür kemik kanseri geliştirme riski biraz artmıştır. RAPADILINO sendromu olan bireylerde, osteosarkom en sık çocukluk veya ergenlik döneminde gelişir ve lenfoma tipik olarak genç erişkinlikte gelişir.
Sahip olduğu isim bozukluğun karakteristik
özelliklerinin kısaltmasıdır: radyal ışın malformasyonları için RA, patella ve
damak anormallikleri için PA, ishal ve çıkık eklemler için DI, ekstremite
anormallikleri ve küçük boyut için LI ve ince burun ve normal zeka için NO. .
Rapadilino sendromunun çeşitli belirtileri ve semptomları, Baller-Gerold sendromu ve Rothmund-Thomson sendromu gibi diğer hastalıkların özellikleri ile örtüşmektedir. Bu sendromlar ayrıca radyal ışın kusurları, iskelet anormallikleri ve yavaş büyüme ile de karakterize edilir. Bu koşulların tümü aynı gendeki mutasyonlardan kaynaklanabilir. Bu benzerliklere dayanarak, araştırmacılar Baller-Gerold sendromu, Rothmund-Thomson sendromu ve Rapadilino sendromunun ayrı bozukluklar mı yoksa örtüşen belirti ve semptomları olan tek bir sendromun parçası mı olduğunu araştırıyorlar.
Sıklık
Rapadilino sendromu nadir görülen bir durum olmasına rağmen dünya çapında prevalansı (yaygınlık) bilinmemektedir. Bu durum ilk olarak, diğer bölgelerde bulunmasına rağmen, 75.000 kişide tahmini 1’i etkilediği Finlandiya’da tespit edildi.
Klinik açıklama
Büyüme gecikmesi hem doğum öncesi hem doğum sonrasıdır. Beslenme problemleri ve bilinen bir nedeni olmayan ishalden dolayı ciddileşir.
Nedenleri
RECQL4 genindeki mutasyonlar Rapadilino sendromuna neden olur. Bu gen, RecQ helikaz denilen bir protein ailesinin bir üyesini yapmak için talimatlar sağlar. Helikazlar, DNA’ya bağlanan ve geçici olarak DNA molekülünün iki spiral şeridini (çift sarmal) çözen enzimlerdir. Bu çözülme, DNA’nın hücre bölünmesine hazırlanırken kopyalanması (çoğaltılması) ve hasarlı DNA’nın onarılması için gereklidir. RECQL4 proteini, vücut hücrelerinde genetik bilginin dengelenmesine yardımcı olur ve DNA’nın kopyalanmasında ve onarılmasında rol oynar.
Rapadilino sendromunda yer alan en yaygın RECQL4 gen mutasyonu, RECQL4 proteininin yanlış biraraya getirilmesine neden olur. Bu genetik değişim, ekzon 7 olarak adlandırılan bir bölge eksik olan ve bir helikaz gibi davranamayan bir proteinin üretilmesine neden olur. Helikaz fonksiyonunun kaybı, normal DNA replikasyonunu ve onarımını önleyebilir ve zaman içinde bir kişinin genetik bilgisinde yaygın hasara neden olabilir. Bu değişiklikler, DNA hatalarının birikmesine ve hücre ölümüne neden olabilir, ancak RECQL4 gen mutasyonlarının Rapadilino sendromunun spesifik özelliklerine neden olduğu kesin değildir.
Kalıtım şekli
Bu durum otozomal resesif bir kalıtsal kalıtımla ifade edilir, yani her bir hücredeki genin her iki kopyası da mutasyonlara sahiptir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak genellikle durumun belirtileri ve semptomlarını göstermezler.
Belirtiler
Çoğu hastalık için semptomlar kişiden kişiye değişecektir. Aynı hastalığı olan insanlar listelenen tüm belirtilere sahip olmayabilir. Bu bilgi İnsan Fenotip Ontolojisi (HPO) olarak adlandırılan bir veri tabanından gelir. HPO, tıbbi kaynaklarda açıklanan belirtiler hakkında bilgi toplar. HPO düzenli olarak güncellenmektedir.
Başparmak yok
Yok/küçük diz kapağı
Göz kapakları arasında dar açıklık
Teşhis
Genetik veya nadir bir hastalık için tanı koymak genellikle zor olabilir. Sağlık uzmanları tipik olarak bir tanı koymak için bir kişinin tıbbi geçmişine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına bakar. Aşağıdaki kaynaklar bu durum için teşhis ve test ile ilgili bilgi sağlar. Teşhis konması hakkında sorularınız varsa, bir sağlık uzmanına başvurmalısınız.
Test Kaynakları
Genetik Test Kayıt Defteri (GTR), bu durum için genetik testler hakkında bilgi sağlar. GTR için hedef kitle, sağlık hizmeti sağlayıcıları ve araştırmacılardır. Genetik test hakkında özel soruları olan hastalar ve tüketiciler bir sağlık kuruluşu veya genetik uzmanı ile irtibata geçmelidir.
Ayırıcı Tanı
Ayırıcı tanılar arasında Rapadilino sendromu ile önemli örtüşmeler gösteren Rothmund-Thomson sendromu (RTS) ve Baller-Gerold sendromu bulunmaktadır. RECQL4 geninin mutasyonları da bu sendromlar için tarif edilmiştir. RTS’nin önemli bir belirtisi olan poikiloderma’nın varlığı, bu sendromu Rapadilino ‘dan ayırır. Rapadilino sendromunda sabit olan radial hipoplazi veya aplaziler RTS’de nadirdir. Aynı şekilde, Baller-Gerold sendromunda kraniyosinozis varlığı, onu Rapadilino sendromundan ayırmaktadır. Üç sendrom, malign patolojilerin gelişmesi riskini belirler, ancak bu RTS (özellikle osteosarkom ve kutanöz kanserler) için daha büyüktür. Klinik farklılıklar fenotip-genotip korelasyonu, özellikle de Rapadilino sendromunda helikazların korunması ile açıklanabilir.
Yönetim ve Tedavi
Gerekirse ortopedik ve beslenme yönetimi önerilmekte ve osteosarcom’u düşündüren işaretler durumunda uygun araştırma önerilmektedir.
Bir Uzman Bulun
Tıbbi yardıma ihtiyacınız olursa, bu hastalık da deneyimi olan doktorları veya diğer sağlık profesyonellerini arayabilirsiniz. Bu uzmanları savunma kuruluşları, klinik deneyler veya tıbbi dergilerde yayınlanan makaleler aracılığıyla bulabilirsiniz. Ayrıca bölgenizdeki bir üniversite veya üçüncül tıp merkezi ile iletişim kurmak isteyebilirsiniz, çünkü bu merkezler daha karmaşık vakalar görmeye, en son teknoloji ve tedavilere sahiptir.
Bölgenizde bir uzman bulamıyorsanız, ulusal
veya uluslararası uzmanlarla iletişim kurmayı deneyin. Sizi, bildiği birisine
konferanslar veya araştırma çalışmaları yoluyla yönlendirebilirler. Bazı
uzmanlar, sizinle ilgilenmek için seyahat edemiyorsanız, telefonla veya e-posta
yoluyla sizinle veya yerel doktorlarınızla görüşmek isteyebilir.
Rett
Sendromu hemen hemen yalnızca kızları etkileyen ilerleyici nörogelişimsel bir
hastalıktır.Sadece nadir vakalarda erkekler etkilenir.Durumun en çok yaygın
formu klasik Rett Sendromudur.Doğumdan sonra,klasik Rett Sendromlu kızlarda dil
ve iletişimde,öğrenmede,koordinasyonda ve diğer beyin fonksiyonlarında şiddetli
problemler gelişmeden önce 6-18 aya kadar görünüşte gelişimleri normaldir.Erken
çocuklukta,etkilenmiş kızlar ellerini amaçlı kullanmayı kaybederler ve
tekrarlayıcı çamaşır sıkma,el yıkama ve el çırpma hareketleri yapmaya
başlarlar.Onlar diğer çocuklara göre daha yavaş büyümeye meyillidirler ve
yaklaşık dörtte üçü küçük kafa boyutuna sahiptir(mikrosefali).Gelişebilecek
diğer işaret ve semptomlar solunum anomalileri,tükürme yada salya akıtma,yoğun
olarak dik dik bakma ve aşırı göz kırpma gibi alışılmadık göz hareketleri,soğuk
eller ve ayaklar,irritabilite,uyku rahatsızlıkları,nöbetler ve omurga
eğriliğinin anormal olarak kayması(skolyoz) içerir.
Araştırmacılar
Rett Sendromunun klasik Rett Sendromundan daha
ılımlı yada şiddetli olabilen çeşitli varyant yada atipik formlarını
tanımlamışlardır.
Rett
Sendromu aynı genetik sebepli spektrum hastalıklarının bir
parçasıdır.Spektrumdaki diğer hastalıklar PPM-X Sendromu,MECP2 Duplikasyon
Sendromu ve MECP2 ile bağlantılı şiddetli neonatal ensefelopatiyi içerir.Bu
diğer durumlar erkekleri de etkileyebilir.
Sıklık
Bu
durum yaklaşık olarak 9.000-10.000 kişiden 1 kızı etkiler.
Sebepleri
MECP2
denilen gendeki mutasyonlar neredeyse tüm klasik Rett Sendromlu olgularda ve bu
durumun diğer formlarının altında yatan sebeptir.Bu gen beyin fonksiyonları
için kritik olan protein yapımı(MeCP2) için talimatlar verir.MeCP2 proteinin
kesin fonksiyonu belirsiz olmasına rağmen,büyük ihtimalle
bağlantılarla(sinapslar) sinir hücreleri(nöronlar) arasında korumayı
kapsar.Ayrıca diğer tip beyin hücrelerinin normal fonksiyonları için gerekli
olabilir.
MeCP2
proteininin beyindeki genlerin aktivitesini düzenlemeye yardımcı olduğu da bir
fikirdir.Bu protein beyin hücrelerindeki tam proteinlerin farklı
versiyonlarının üretilmesini kontrol edebilir.MECP2 genindeki mutasyonlar MeCP2
proteinini değiştirir ya da daha az protein üretiminin sonucu olarak beyindeki
diğer hücrelerin yada nöronların normal fonksiyonlarının aksaması görünür.Özellikle
çalışmalar öneriyor ki MeCP2 proteini
belirli nöronların aktivitelerini azaltabilir ve diğer bir başkasıyla iletişim
yeteneklerini bozabilir.Bu değişimlerin Rett Sendromunun spesifik özelliklerini
nasıl yönlendirdiği belirsizdir.
Kalıtım Paterni
Rett
Sendromlu insanların %99’undan fazlasının ailelerinde hastalık hikayesi
yoktur.Bu olguların çoğu MECP2 genindeki yeni mutasyonlar sonucunda olur.
Birden
fazla etkilenmiş aile üyesi olan bazı aileler tanımlanmıştır.Bu olgular MECP2
genindeki mutasyonlar sebebiyle olmuş klasik Rett Sendromu ve varyantlarının
araştırmacılar tarafından tanımlanmasına yardım etmiştir,kalıtım paterni X’e
bağlı dominanttır.Eğer X kromozomu üzerine konumlanmış mutasyona uğramış gen
hastalığa sebep oluyorsa,iki cinsiyet kromozomundan bir tanesi,durum
değerlendirilmelidir.Eğer değişmiş genin bir kopyası her hücrede bu duruma
sebep olmak için elverişliyse kalıtım dominanttır.
MECP2
geninde mutasyonlar olan erkekler sıklıkla çocukken ölürler.Yinede MECP2
geninde değişiklik içeren az sayıda erkekte Rett Sendromuna benzer işaret ve
semptomlar gelişebilir,bunlar entellektüel disabiliteyi,nöbetleri ve hareket
problemlerini kapsar.Erkeklerde bu durum MECP2 ile ilişkili şiddetli neonatal
ensefalopati olarak tanımlanmıştır.MECP2 genindeki mutasyona sahip bazı
erkeklerde işaret ve serkeklerde spektrumun sonunda daha ılımlıdır.
Teşhis
Rett
Sendromunun teşhisi karakteristik semptomların tanımına,detaylı hasta
hikayesine ve klinik değerlendirmeye dayanmaktadır.Çeşitli özelleşmiş testler
benzer semptomlara sebep olan diğer durumların ekarte edilmesine yol
gösterebilir.Son zamanlarda belirlenmiş güncellenen teşhis kriteri
yayınlanmıştır.(2010)Bu teşhis kriterinin tamamlanması Rett Sendromunun klinik
teşhisine götürebilir.Bu rapor ayrıca Rett Sendromunun çeşitli formlarını
kapsar.Moleküler genetik testleme MECP2 genindeki mutasyonların varlığını
belirler ve Rett Sendromunun klinik teşhisini doğrular.
Standart Terapiler
Tedavi
Rett
Sendromunun tedavisi her bireyde görülen spesifik semptomlar doğrultusunda
yönlendirilir.Tedavi uzmanlardan oluşmuş bir takımın koordinasyonu ve gayretine
gerek duyabilir.Çocuk doktorları,çocuk nörologları,gastroentereologlar,konuşma
terapistleri,psikiatrlar,diyetisyenler ve diğer sağlık profesyonellerinin
etkilenmiş çocuğun tedavisini sistematik ve kapsamlı olarak planlamaya
ihtiyaçları vardır.Genetik danışma etkilenmiş bireyler ve aileleri için faydalı
olabilir.
Rett
Sendromlu bireylerin tedavi edilmesi için tedavi çeşitleri kompleks ve
çeşitlidir.Spesifik tedavi planının son derece bireyselleştirilmiş olmaya
gereksinimi vardır.Etkilenmiş çocuğun potansiyeline erişebilmeyi garanti etmek
için erken gelişimsel müdahale önemlidir.Etkilenmiş çocukların çoğu
mesleki,fiziksel ve konuşma terapisinden yararlanırlar.Rehabilitatif ve
davranışsal terapinin çeşitli metotları
faydalı olabilir.İlaveten tedavi edici eğitimi içeren medikal,sosyal ve/veya
mesleki hizmetler gerekli olabilir.Tüm aile için psikososyal destek temeldir.
Diğer
tedavi semptomatik ve sistematiktir.Rett Sendromu için ek terapiler spesifik
anomalilerin ortaya çıkışına ve genellikle standart rehberleri takip etmeye
bağlıdır.
Semptomlar
Apraksi
Disfaji
EEG’de anormallik
Entelektüel
disabilite(şiddetli)-Erken ve şiddetli mental retardasyon
Mikrosefali(Anormal derecede
küçük kafatası)
Rett Sendromunun Diğer İsimleri(Kısaltmaları)
Klasik Rett Sendromu
RTT
Varyant(Atipik) Rett Sendromu
Rett Hastalığı
Rett’in Hastalığı
Otizm-Demans-Ataksi-Amaçlı el
kullanımının kaybı Sendromu
Musa Çelik- Acıbadem
Üniversitesi, Moleküler Biyoloji ve Genetik
Genel Bilgi
Araknoid (örümceksi zar, beyin zarlarından biri)
kistler (içi koloit, yağ vb. sıvı
veya yarı sıvı bir madde ile dolu 20 mm’den küçük patolojik torba, kitle)1, beynin yüzeyi ile
beyin omuriliğini örten üç beyin zarı (meningeal) tabakasından biri olan,
beynin yüzeyi ile kafatası (kranyal) tabanı arasında veya araknoid zar üzerinde
gelişebilen, araknoidal hücreler ve kollajen2 ile kaplı beyin omurilik
sıvısıdır. Araknoid kistler doğuştan bir hastalıktır3 ve çoğu vaka
bebeklik döneminde başlar; ancak, ergenliğe kadar gecikme olabilir.4
Genetik Değişiklikler/Etken Faktörler
Araknoid kistlerin kesin nedeni
bilinmemektedir. Araştırmacılar, çoğu araknoid kist vakasının, araknoid zarın
açıklanamayan bir şekilde bölünmesinden veya yırtılmasından kaynaklanan
gelişimsel şekil bozukluklarından (malformasyon)
olduğunu düşünmektedir.
Bazı durumlarda, orta fossada (kafa orta çukuru) meydana gelen araknoid
kistlere, az gelişmişlik (hipoplazi)
veya temporal lobun (insan beynindeki 4
beyin lobundan biri) sıkışması eşlik eder. Temporal lob anormalliklerinin
orta fossa araknoid kistlerin gelişiminde oynadığı rol hakkında kesin bir bilgi
yoktur.
Kalıtsal bozuklukların araknoid kistlerle
bağlantılı olduğu bazı durumlar vardır.5
Minör kafa travması nedeniyle bir kist zarar
gördüğünde, araknoid kistlerin bazı etkileri (komplikasyonları) ortaya çıkabilir.6 Travma, bir kist
içindeki sıvının diğer alanlara (örneğin
subaraknoid boşluk) sızmasına neden olabilir. Bir kistin yüzeyindeki kan
damarları yırtılarak (intrasistik kanama)
kistin büyüklüğünü artırabilir. Bir kan damarı kistin dışında kanarsa, kan
toplanması (hematom) oluşabilir. İntrasistik
kanama ve hematom vakalarında, birey kafatası içinde artan basınç semptomlarına
ve yakındaki sinir dokusunun sıkışması belirtilerine sahip olabilir.
Bazı bilim insanları araknoid kistlerin gerçek bir kongenit (doğuştan) durumu olup olmadığını veya bunun ikincil kistlerden ayrılması gerekip gerekmediğini tartışır.7 Son zamanlarda yapılan bir çalışmada CT (bilgisayarlı tomografi) sisternografi ile araknoid kist ve subaraknoid boşluk arasındaki iletişimde farklılıklar olduğunu göstermektedir.8 Bir dizi hastada araknoid kist sıvısı ve BOS’un karşılaştırılması sonucu kimyasal bileşiminde farklılıklar olduğu görülmüştür.9
Resim 1: Çocukluktan beri nöbet öyküsü olan 26 yaşındaki bir erkeğin kafasında
yapılan artmış BT taraması (bir sonraki resimde olduğu gibi). Tarama kitle
etkisi ile büyük bir sol frontoparietal kist gösterir. Kisten parça alındı ve
histolojik analiz araknoid kisti 25
Belirtiler
Çoğu durumda, araknoid kistler kongenit
bulunur, ancak genellikle bir bireyin yaşamı boyunca herhangi bir semptoma
neden olmaz. Daha az sıklıkla, kafa travması, tümör varlığı, enfeksiyon veya
beyinde ameliyat nedeniyle araknoid kistler gelişebilir.
Bazı durumlarda araknoid kistler belirtilere
neden olmaz.10 Kistin her bireydeki yeri ve büyüklüğü belitilerin
ortaya çıkıp çıkmadığını ve ne zaman başlayabileceklerini belirlemeye yardımcı
olur. Çoğu bireyde 20 yaşından önce ve özellikle doğumdan sonraki ilk yıllarda belirtiler
gelişir.10,11
Araknoid kistlerin belirtileri
beynin neresinde bulunduklarına bağlı olarak değişir. 10,11
Baş
ağrısı,
Mide
bulantısı,
Kusma,
Beyinde
aşırı beyin omurilik sıvısı birikmesi (hidrosefali), kafa içi basıncının
artmasına neden olur,
Kafatası
kemiklerinin şekil bozuklukları,
Nöbetler,
İşitme
ve görme bozuklukları,
Baş
dönmesi,
Denge
ve yürüme zorluğu,
Gelişimsel
gecikmeler, davranış değişiklikleri, istemli hareketleri kontrol edememe
(ataksi), bilişsel bozukluk ve vücudun bir tarafında zayıflık veya felç gibi
nörolojik bulgular (hemiparezi).
Omuriliğin etrafındaki araknoid kistler,
omuriliği veya sinir köklerini sıkıştırabilir ve ilerleyici sırt ve bacak
ağrısı, bacaklarda veya kollarda karıncalanma veya uyuşukluk ve zayıflık gibi belirtilere
neden olabilir. Bazı durumlarda, idrar veya dışkı tutamama gibi sorunlara neden
olabilir.10,11,12
Genetik Görülme Sıklığı
Araknoid kistler popülasyonun % 1,1’inde
görülür13,14 cinsiyet dağılımı 2: 1 erkek: kadın.15
Bunların sadece% 20’sinde belirtiler vardır, bunlar genellikle ikincil hidrosefalidir
(beyin omurilik sıvısının çoğalmasıyla, beyin karıncıklarının, kimi zaman da
kafatasının büyümesine yol açan bir hastalık).13
2,536 sağlıklı genç erkeğin incelendiği bir çalışmada%
1,7 (% 95 CI% 1,2 – 2,3) prevalans (yaygınlık) görülmüştür. Tespit edilen
anormalliklerin yalnızca küçük bir yüzdesi acil tıbbi yardım gerektirmektedir.16
Kalıtım Paterni/Deseni
Araknoid kistlerin tanısı konulan olguların
çoğu sporadiktir (ailede araknoid kist öyküsü olmayan kişilerde görülür).17
Bununla birlikte, tıbbi literatürde ailesel araknoid kist vakalarına da
rastlanmıştır, bu da genetik yatkınlığın bazı insanlarda rol oynayabileceğini
düşündürmektedir.10 Ailesel olgularda kalıtım otozomal resesif olabilir.17,18
Teşhis Yöntemleri
Araknoid kistlerin
teşhisi genellikle nöbet geçiren bir kişinin muayenesi sırasında tesadüfi
olarak yapılır. Ayrıntılı bir hasta öyküsü, ayrıntılı bir klinik muayene ve
çeşitli uzmanlık testleri, özellikle bilgisayarlı tomografi (BT taraması) ve
manyetik rezonans görüntüleme (MRI) gibi ileri görüntüleme teknikleri
yardımıyla bir teşhis şüphesi olabilir. BT taramaları ve MRI’leri araknoid
kistlerin varlığını ortaya çıkarabilir. BT taraması sırasında, beynin doku
yapısının kesit görüntülerini gösteren bir film oluşturmak için bir bilgisayar
ve X ışınları kullanılır. MRI sırasında beynin kesitsel görüntülerini
oluşturmak için bir manyetik alan ve radyo dalgaları kullanılır.
Tedaviler
Araknoid kistlerin
çoğu tesadüfen bulunur ve yok olmaz. Bu sebeple hekimler konservatif (koruyucu)
tedavi önerirler. Bireyde belirtiye rastlanmadığında, tedaviye gerek
kalmayabilir ve etkilenen bireyler periyodik olarak gözlem altında tutulur.
Belirtiler ortaya çıkarsa, tedavi için yeniden değerlendirme yapılır.
Tedavi gerek duyulursa,
uygulanacak spesifik (özel) tedavi semptomların mevcut olup olmadığına, kistin
büyüklüğüne ve kistin kafatasındaki spesifik konumuna bağlıdır.
Tedavinin
önerildiği durumlarda, tedavi geleneksel olarak iki işlemden oluşur. Bunlar
kafatasında delik açma (an
open craniotomy fenestration) ve ventriküloperitoneal
şant uygulaması (ventriculoperitoneal shunting).
Bir kafatasında
delik açma uygulaması, beyne ve omuriliğe ait (cerebrospinal) sıvının yeniden
emildiği araknoid zarın altındaki (subaraknoid) boşluğa akmasını sağlamak için
kist duvarında çok sayıda açıklık oluşturulmasıdır (fenestrasyonlar). Alternatif
olarak, bazı durumlar ya beynin ventriküler sistemine ya da karın boşluğuna
drenaj sağlamak için kistin içine bir cihazın (şant) cerrahi olarak
sokulmasıyla tedavi edilebilir. Bu, kisti boşaltacak ve beyin omurilik
sıvısının dolaşması için yeterli bir geçiş yolu sağlayacaktır. 19
Beyin şantının cerrahi olarak yerleştirilmesi20
Fenestrasyon (Delik Açma)
İğne aspirasyonu veya trepanasyon
Kapsül rezeksiyonu21
Farmakolojik tedaviler, nöbet veya ağrı gibi
spesifik semptomları ele alabilir.
Sınıflandırma
Araknoid
kistler beyinde veya omurgada bulunur. Kafatası-içi (inrtakraniyal) araknoid
kistler genellikle araknoidal sarnığa bitişik olarak ortaya çıkar. 22
Spinal araknoid kistler ekstradural, intradural veya perineural olabilir ve
radikülopatiye işaret eden belirti ve semptomlarla ortaya çıkma eğilimindedir.22
Araknoid
kistler ayrıca primer (doğuştan) veya sekonder (edinilmiş) olarak
sınıflandırılabilir ve insanlarda, kedilerde ve köpeklerde rapor edilmiştir.23
Araknoid
kistler nispeten asemptomatik olabilir veya sinsi belirtilerle ortaya
çıkabilir; bu nedenle tanı sıklıkla gecikir.
Hastalıkla İlişkili Genler
Araknoid kistlerle ilişkili
olduğu düşünülen iki tane gen vardır:24
^ Ariai S, Koerbel A, Bornemann A, Morgala M,
Tatagiba M (2005). “Cerebellopontine angle arachnoid cyst harbouring
ectopic neuroglia”. Pediatr Neurosurg. 41 (4): 220–3.
doi:10.1159/000086566. PMID 16088260.
^ Gelabert-González M (2004).
“Intracranial arachnoid cysts]”. Rev Neurol (in Spanish). 39 (12):
1161–6. PMID 15625636.
^ Jump up to:a b c d e f g h i j k l m
“Arachnoid Cysts Information Page”. NINDS. Retrieved April 7, 2017.
^ Schievink WI, Huston J, Torres VE, Marsh WR
(December 1995). “Intracranial cysts in autosomal dominant polycystic
kidney disease”. J. Neurosurg. 83 (6): 1004–7.
doi:10.3171/jns.1995.83.6.1004. PMID 7490613.
^ De K, Berry K, Denniston S (July 2002).
“Haemorrhage into an arachnoid cyst: a serious complication of minor head
trauma”. Emerg Med J. 19 (4): 365–6. doi:10.1136/emj.19.4.365. PMC
1725893. PMID 12101165.
^ Westermaier T, Schweitzer T, Ernestus RI
(2012). “Arachnoid cysts”. Adv. Exp. Med. Biol. Advances in
Experimental Medicine and Biology. 724: 37–50. doi:10.1007/978-1-4614-0653-2_3.
ISBN 978-1-4614-0652-5. PMID 22411232.
^ Wang X, Chen JX, You C, Jiang S (July 2012).
“CT cisternography in intracranial symptomatic arachnoid cysts:
classification and treatment”. J. Neurol. Sci. 318 (1–2): 125–30.
doi:10.1016/j.jns.2012.03.008. PMID 22520095.
^ Berle M, Wester KG, Ulvik RJ, Kroksveen AC,
Haaland OA, Amiry-Moghaddam M, Berven FS, Helland CA (June 2010).
“Arachnoid cysts do not contain cerebrospinal fluid: A comparative
chemical analysis of arachnoid cyst fluid and cerebrospinal fluid in
adults”. Cerebrospinal Fluid Res. 7: 8. doi:10.1186/1743-8454-7-8. PMC
2898803. PMID 20537169.
Arachnoid Cysts. National Organization for
Rare Disorders (NORD). 2015; http://rarediseases.org/rare-diseases/arachnoid-cysts/.
NINDS Arachnoid Cysts Information Page. NINDS.
September 11, 2015;
https://www.ninds.nih.gov/Disorders/All-Disorders/Arachnoid-Cysts-Information-Page.
Petridis AK, Doukas A, Barth H & Mehdorn
HM. Spinal cord compression caused by idiopathic intradural arachnoid cysts of
the spine: review of the literature and illustrated case. European Spine
Journal. 2010; 19(2):124-129.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2899629/.
^ Jump up to:a b Flaherty AW. The
Massachusetts General Hospital Handbook of Neurology 2000 Jan 1;105. (ISBN
0-683-30576-X)
^ Vernooij MW, Ikram MA, Tanghe HL, Vincent
AJ, Hofman A, Krestin GP, Niessen WJ, Breteler MM, van der Lugt A (November
2007). “Incidental findings on brain MRI in the general population”.
N. Engl. J. Med. 357 (18): 1821–8. doi:10.1056/NEJMoa070972. PMID 17978290.
^ Pradilla G, Jallo G (February 2007).
“Arachnoid cysts: case series and review of the literature”.
Neurosurg Focus. 22 (2): E7. PMID 17608350.
^ Weber F, Knopf H (January 2006).
“Incidental findings in magnetic resonance imaging of the brains of
healthy young men”. J. Neurol. Sci. 240 (1–2): 81–4.
doi:10.1016/j.jns.2005.09.008. PMID 16256141.
Cassandra L. Kniffin. Arachnoid Cysts, Intracranial. Online Mendelian
Inheritance in Man (OMIM). December, 2007; http://omim.org/entry/207790.
Cincu R, Agrawal A, Eiras J. Intracranial arachnoid cysts: current concepts
and treatment alternatives. Clin Neurol Neurosurg. 2007;109:837-843.
^ Strojnik T (2006). “Different
approaches to surgical treatment of arachnoid cysts”. Wien. Klin.
Wochenschr. 118 Suppl 2: 85–8. doi:10.1007/s00508-006-0540-2. PMID 16817052.
^ Jump up to:a b Yamakawa H, Ohkuma A, Hattori
T, Niikawa S, Kobayashi H. “Primary intracranial arachnoid cyst in the
elderly: a survey on 39 cases”, Acta Neurochir (Wien). 1991;113(1-2):42-7.
(PMID 1799142)
^ Jump up to:a b Arachnoid cyst. (n.d.). Gale
Encyclopedia of Neurological Disorders. Retrieved September 10, 2006, from
Answers.com Web site: http://www.answers.com/topic/arachnoid-cyst
^ Reed SD, Cho DY, Paulsen D. Quadrigeminal
Arachnoid Cysts in a kitten and a dog. J Vet Diagn Invest 2009. 21(5):707-710.
Danon
hastalığı, kalp kasının ve hareket kaslarının zayıflamasına (kardiyomiyopathi
ve miyopathi) ve zihinsel engelliliğe sebep olan nadir bir genetik hastalıktır.
Bu hastalık bir tür lizozom deposu hastalığıdır. Danon hastalığında, lizozom
zarında bozukluk vardır. Erkek bireylerde hastalığın belirtileri daha erken görülür
ve kadınlara oranla daha ciddi olur. Belirti ve semptomlar erkeklerde çocukluk
çağında, kadınlarda erken yetişkinlik döneminde ortaya çıkar. Etkilenen
erkekler ortalama 19 yaşına kadar, etkilenen kadınlar ortalama 34 yaşında kadar
yaşar.
Genetik Değişiklikler/Etken Faktörler
Danon
hastalığına LAMP2 genindeki mutasyon sebep olur. LAMP2 geni, lizozomda bulunan
LAMP-2 proteinini yapmada görev alır. Lizozomlar, hücre içi sindirimde rol
alır. LAMP-2 proteinin lizozomdaki rolü hala belli değildir. LAMP2 genindeki
mutasyonlar LAMP-2 proteinin üretiminde azalmaya sebep olur bu da hücresel
materyallerin lizozoma taşınma sürecine zarar verir. Bazı araştırmalara göre
LAMP-2 proteini olmayan hücrelerde otofajik vakuol birikimine sebep olunabilir.
Danon hastalığına sahip bireylerin kas hücrelerinde anormal büyük otofajik
vakuol olabilir. Bu birikim kas hücrelerinin çalışmamasına sebep olabilir ve
kas zayıflığı görülebilir.
Belirti ve Semptomlar
Erkeklerde
kadınlara göre belirti ve semptomlar daha erken ortaya çıkar ve daha ciddi
olabilir. Erkeklerde semptomlar çocukluk çağında ortaya çıkarken kadınlarda
erken yetişkinliğe kadar belirtiler ortaya çıkmayabilir. Kardiyomiyotapi (kalp
kası bozukluğu), en sık görülen semptomdur ve hastalıktan etkilenmiş tüm
erkeklerde görülür. Kardiyomiyopatinin görüldüğü kadınların yarısında
hipertropik kardiyomiyopati (kalp kasının kalınlaşması) ve diğer yarısında
dilate kardiyomiyopati (kalbin genişlemesi ve zayıflaması) görülür. Çoğu
erkekte hipertropik kardiyomiyopati daha fazla görülür. Bu iki kalp sorunu da
kalp yetmezliği sebebiyle ölümcül olabilir. İskeletsel miyopati (hareket
kaslarının hastalığı), çoğu erkekte ve kadınların yaklaşık yarısında görülür.
Kaslardaki zayıflık genelde üst kollarda, omuzlarda ve boyunda olur. Etkilenmiş
bireyler de düzensiz kalp atışı veya göğüs ağrısı gibi kalbe bağlı başka belirtiler
ortaya çıkabilir. Bazılarında kalp atışını kontrol eden elektrik sinyallerinde
anormalleşe de görünebilir. Çoğu erkekte zeka geriliği görünürken kadınlarda
normal zeka gelişimi görülür. Yaygın olmayan ve daha az görülen belirtiler:
mide-bağırsak hastalığı, nefes alma problemleri ve görüş anomalileri.
Genetik Görülme Sıklığı
Bu hastalığın görülme
sıklığı tam olarak bilinmemektedir ama erkekler bu X kromozomuna bağlı dominant
hastalıktan daha ciddi bir şekilde etkilenmektedir.
Kalıtım Paterni/Deseni
Bu hastalık X
kromozomuna bağlı dominant olarak olarak kalıtılmaktadır. Bu tip kalıtsallıkta
erkeklerde kadınlara göre belirti ve semptomları daha erken yaşta ortaya çıkar.
Teşhis Yöntemleri ve Tedaviler
Teşhis için
genetik test yaptırılabilir. Tedavi için semptomlar gözden geçirilir ve
kardiyolog, nörolog, genetik danışmanı, fizik tedavi uzmanı gibi bir ekipten
yardım alınabilir. Danon hastalığı hızlı ilerleyen kardiyomiyopatiye ve ani
ölüme sebep olduğu için kalp hastalığını düzenli kontrol ettirmek önemlidir.
Fizik tedavi ile de kas gücünün sürdürülmesine yardımcı olunabilir. Zeka
geriliği görülen erkeklerde erken eğitim müdahelesi gerekebilir.
Hastalıkla İlişkili Genler
LAMP2
genindeki mutasyon, Danon hastalığına sebep olur.
Hastalığın Diğer İsimleri
Antopol
disease, glycogen storage cardiomyopathy, glycogen storage disease type IIB,
GSD IIB, lysosomal glycogen storage disease without acid maltase deficiency,
pseudoglycogenosis II, vacuolar cardiomyopathy and myopathy X-linked
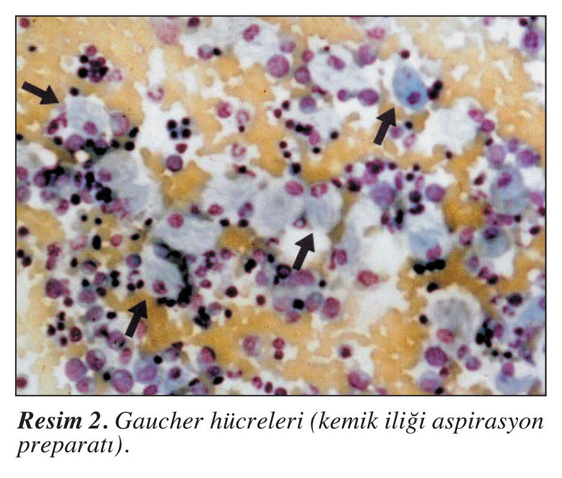
Gaucher hastalığı, vücudun birçok organını
ve dokusunu etkileyen kalıtsal bir hastalıktır. Bu durumun belirti ve
semptomları etkilenen bireyler arasında geniş ölçüde değişir. Araştırmacılar,
karakteristik özelliklerine dayanarak birçok Gaucher hastalığı türü
tanımlamışlardır. Gaucher hastalığının yaygın belirtileri
arasında anormal şekilde genişletilmiş bir karaciğer ve / veya dalak
(hepatosplenomegali), düşük dolaşımdaki kırmızı kan hücreleri (anemi), düşük
trombosit (trombositopeni) seviyeleri ve iskelet anomalileri bulunur.
Trombositler pıhtılaşmayı destekleyen kan hücreleridir ve trombositopenisi olan
hastalarda kanama problemleri ortaya çıkabilir. Üç ayrı Gaucher hastalığı şekli
tanımlandı ve nörolojik komplikasyonların yokluğu veya varlığı ve kapsamı ile
ayırt edildi. Gaucher hastalığının üç formu da otozomal resesif paternde
kalıtsaldır. [
Tip 1 Gaucher hastalığı bu durumun en sık
görülen şeklidir. Tip 1 ayrıca nöronopatik olmayan Gaucher hastalığı olarak da
adlandırılır çünkü beyin ve omurilik (merkezi sinir sistemi) genellikle
etkilenmez. Bu durumun özellikleri hafif ila şiddetli arasında değişmektedir ve
çocukluktan yetişkinliğe kadar her zaman ortaya çıkabilir. Başlıca belirti ve
semptomlar arasında karaciğer ve dalak (hepatosplenomegali), az sayıda kırmızı
kan hücresi (anemi), kan plateletlerinde (trombositopeni), akciğer hastalığında
ve kemik ağrısı gibi kemik anormalliklerinde bir azalmanın neden olduğu kolay
morarma, kırıklar ve artrit.
Tip 2 ve 3 Gaucher hastalığı, hastalığın
nöronopatik formları olarak bilinir, çünkü merkezi sinir sistemini etkileyen
problemlerle karakterize edilir. Yukarıda açıklanan belirti ve semptomlara ek
olarak, bu durumlar anormal göz hareketlerine, nöbetlere ve beyin hasarına
neden olabilir. Tip 2 Gaucher hastalığı genellikle bebeklikten itibaren hayatı
tehdit eden tıbbi sorunlara neden olur. Tip 3 Gaucher hastalığı ayrıca sinir
sistemini de etkiler, ancak tip 2’den daha yavaş kötüleşme eğilimindedir.
En şiddetli Gaucher hastalığı tipi
perinatal ölümcül form olarak adlandırılır. Bu durum doğumdan önce veya
bebeklik döneminde başlayan ciddi veya hayati tehlike oluşturan
komplikasyonlara neden olur. Perinatal ölümcül formun özellikleri, doğumdan
önce sıvı birikiminin neden olduğu geniş şişmeyi içerebilir (hidrops fetalis);
kuru, pullu cilt (iktiyoz) veya diğer cilt anormallikleri; hepatosplenomegali;
ayırt edici yüz özellikleri; ve ciddi nörolojik problemler. Adından da
anlaşılacağı gibi, perinatal ölümcül Gaucher hastalığına sahip bebeklerin çoğu
doğumdan sonra sadece birkaç gün hayatta kalır.
Görülme Sıklığı
Gaucher hastalığı, genel popülasyondaki 50.000 ila 100.000 kişiden 1’inde görülür. Tip 1, hastalığın en yaygın şeklidir; Aşkenazi halklarında (doğu ve orta Avrupa) Yahudi mirası, diğer kökenlere göre daha sık görülür. Bu koşul biçimi, Aşkenazi Yahudi mirasının 500 ila 1000 kişiden birini etkilemektedir. Gaucher hastalığının diğer formları nadirdir ve Aşkenazi Yahudi kökenli insanlarda daha sık görülmez.
Oluşumuna Sebep Olan Ana Etkenler
Gaucher hastalığına GBA genindeki
değişiklikler (mutasyonlar) neden olur.
Gaucher hastalığının üç formu da otozomal
resesif paternde kalıtsaldır. Resesif genetik bozukluklar, bir
birey her bir ebeveynden anormal bir gen aldığında ortaya çıkar. Bir birey her
bir ebeveynden hastalık için bir normal gen ve bir anormal gen alırsa, kişi
hastalık için taşıyıcı olacaktır, ancak genellikle semptom göstermez. İki
taşıyıcı ebeveynin her ikisinin de anormal geni geçmesi ve bu nedenle,
etkilenen bir çocuğa sahip olma riski her hamilelikte% 25’tir. Ebeveynler gibi
taşıyıcı olan bir çocuğa sahip olma riski her hamilelikte% 50’dir. Bir çocuğun
her iki ebeveynden normal gen alma şansı% 25’tir. Risk erkeklerde ve kadınlarda
aynıdır.
Teşhis
Kan testleri, düşük seviyelerde
glikoserebrosidaz tanımlayabilir.
Genetik testler en yaygın dört genetik
mutasyon olan N370S, L444P, 84gg ve IVS2 [+1] ve bazı daha az yaygın olanları
arar. Bu test tam olarak güvenilir değildir, çünkü Gaucher hastalığı ile
bağlantılı olabilecek tüm genetik mutasyonlar henüz bilinmemektedir.
Tedavi
Gaucher hastalığının bilinen etkili bir tedavisi
yoktur, ancak tedavi semptomları azaltmaya ve yaşam kalitesini arttırmaya
yardımcı olabilir.
Tip 2 Gaucher hastalığı olan hastalar için
enzim replasman tedavisi mevcuttur, ancak ortaya çıkması muhtemel olan ciddi
beyin hasarı için etkili bir tedavi yoktur.
Hafif tip 1 olan bazı hastaların tedaviye
ihtiyacı olmayabilir, ancak periyodik olarak izlenmeleri gerekir.
Tip 1 veya 3 Gaucher hastalığı olan
hastalar için, çeşitli tedaviler yardımcı olabilir.
Pinta, ; Küba ve Amerika’nın Güney kısımlarında nadir görülen, treponema carateum bakterisinin neden olduğu ve ciltte tuhaf renk değişiklikleriyle vasıflı olan bulaşıcı bir tropikal hastalıktır. Pinta, çeşitli cilt lezyonları ve renk değişikliği ile karakterize üç farklı aşamada ilerler. Diğer organ sistemleri etkilenmez. Yüz ve ekstremiteler gibi cildin açıkta kalan bölgeleri en sık etkilenir.
Pinta, bir treponemadan kaynaklanan
bulaşıcı bir hastalık olan bir treponematosis olarak
sınıflandırılır. Treponemas, spiral şekilli bakteri cinsidir (spiroketler)
Treponemas, pinta, yaws ve sifiliz, frengi gibi birçok bulaşıcı hastalığa neden
olmaktadır. Zührevi
sifiliz cinsel temasla bulaşır; diğer hastalıklar ise yakın olmayan
nonerereal temasla bulaşır.
Genetik Değişiklikler / Etken Faktörler
Bu hastalığın farklı evreleri açıkça ayrılmaz ve
tezahürlerin örtüşmesi sık görülür. Teşhis
coğrafi bölgeye, klinik bulgulara, eksüdalardaki organizmaların gösterilmesine
ve pozitif serolojiye dayanır. Penisilin tercih edilen
antibiyotiktir. Antibiyotik tedavisinin ardından lezyonların hızla
iyileştiği sifiliz ve sazların aksine, pinta lezyonlarının tamamen çözülmesi
için 1 yıl gerekebilir. Birincil veya erken ikincil belirtilerden sonra,
cilt pigmentasyonu normale döner. Ancak daha sonraki tezahürlerde,
pigmentasyon kalıcı olarak değişmeden kalır.
Belirti ve Semptomlar
Pinta’nın semptomları ve ilerlemesi etkilenen
bireyler arasında değişebilir. Pinta genellikle üç ayrı aşamada ilerler:
ilk lezyonların erken evresi; yaygın (yayılmış) lezyonları olan bir ara
evre; ve geç bir aşama. Kuluçka süresi yedi ila 21 gün arasında değişebilir.
Çoğu durumda, ilk lezyonlar (primer) kolların ve
bacakların açıkta kalan bölgelerinde en sık görülen küçük, kırmızımsı
(eritemli) lekelerdir (papüller). Yüz, boyun, göğüs ve karın da
etkilenebilir. Papüller genellikle kaşıntılıdır (kaşıntılı) ve büyük
plaklar oluşturmak için bir araya gelerek yayılabilir. Bazı durumlarda,
yakındaki lenf düğümleri iltihaplanabilir (lenfadenit).
İlk lezyonların gelişmesinden bir ay ila bir yıl
sonra, etkilenen bireyler pintid denilen ikincil cilt döküntüleri geliştirebilir. Pintidler,
normalde primer lezyonlarla aynı bölgeleri etkileyen küçük pullu, kırmızımsı
lezyonlardır. Kuru ve kabuklu olabilirler (psoriatik pintitler).
Üç aydan bir yıla kadar her yerde, ikincil
lezyonlar ve bazı durumlarda primer lezyonlar rengi yavaşça kırmızıdan
kahverengiye veya kayrak mavisine değiştirebilir. Bu lezyonlar nihayetinde
renklerini kaybedebilir (depigmentasyon) beyazlaşarak ciltte benekli bir
görünüm bırakır. Pintidler 10 yıla kadar tekrarlayabilir.
Pintaların geç fazı, ilk lezyonların
gelişmesinden yaklaşık iki ila beş yıl sonra ortaya çıkar ve beyaz veya renksiz
(akromatik) lezyonlarla karakterize edilir. Bu evrede, etkilenen bireyler
ayrıca ayak tabanları ve ellerin avuçlarında (hiperkeratoz) alışılmadık şekilde
kuru, kalınlaşmış cilt de geliştirebilirler. Sonunda, etkilenen bireyler
bazı bölgelerde kuru, kırışmış ince (atrofik) cilt geliştirebilir.
Genetik Görülme Sıklığı
Pinta erkekleri ve dişileri eşit sayıda
etkiler. Vakaların çoğu, dünyanın endemik bölgelerinden gelen çocuklar
veya gençlerdir. Meksika, Orta Amerika ve Kolombiya gibi güney kırsal
bölgelerinde en yaygın olanıdır. Çeşitli Karayip Adaları’nda düşük
insidans oranları ile ortaya çıkar. Son yıllarda, tıp literatüründe her yıl
sadece birkaç yüz pinta vakası bildirilmiştir. Pinta, etkilenen bireyler
endemik bölgeleri ziyaret etmediği sürece Amerika Birleşik Devletleri’nde
oluşmaz.
1950’lerde ve 60’larda Dünya Sağlık Örgütü (WHO)
tarafından penisilin ile yapılan toplu tedavi kampanyasının ardından pinta
prevalansı büyük ölçüde azaldı. Pinta’nın mevcut insidansı
bilinmemektedir.
Son zamanlarda toplam vaka sayısının 500.000
olduğu tahmin ediliyor. İletim, insandan insana cinsel olmayan temas
yoluyla gerçekleşir. Çoğu vaka başlangıçta çocuklarda ve ergenlerde
görülür.
Teşhis Yöntemleri ve Tedavileri
Pinta, Treponema carateum olarak bilinen spiral
şeklindeki bakterinin (spirochete) neden olduğu bulaşıcı bir tropikal
hastalıktır.
Pinta teşhisi, ayrıntılı bir hasta öyküsü (örneğin, endemik alana son yolculuk), kapsamlı bir klinik değerlendirme, karakteristik semptomların tanımlanması ve çeşitli testlere dayanılarak şüphelenilir. Bu testler, etkilenen bireylerin cilt lezyonlarından alınan doku örneklerinin mikroskopik incelemesini (karanlık alan muayenesi) içerebilir. Diğer kan testleri (örneğin, VDRL ve Treponemal antikor emilim testi [FTA-ABS]) genellikle ikincil cilt lezyonları göründükten sonra pozitif hale gelir.
Tedavi
Pinta tedavisi, antimikrobiyal ilaçları
içerir. Tercih edilen ilaç, benzatin penisilin G’dir. Bu antibiyotiğin tek
bir büyük dozu genellikle cilt lezyonlarını iyileştirir ve organizmayı elimine
eder. Birincil ve ikincil lezyonlar sıklıkla altı ila 12 ay içinde
iyileşir; geç evre lezyonlarının çözülmesi daha fazla zaman
alır. Penisiline alerjisi olan kişiler tetrasiklin veya eritromisin ile
tedavi edilebilir.
İlaç tedavisi, aile bireylerinde ve etkilenen
bireylerle sık temas halinde olan diğerlerinde hastalığın önlenmesi için de
kullanılabilir.
Tietz sendromu, işitme kaybına,
açık renk ten ve saç rengine sebep olan nadir bir hastalıktır. İşitme kaybına
iç kulaktaki anomalilik (sensörinöral işitme kaybı) sebep olmaktadır ve
doğumdan itibaren hastada mevcuttur. Bu hastalığa sahip olan insanlar beyaz saç
ve solgun cilt ile doğarlar ama saç renkleri zamanla koyulaşabilir, göz
renkleri mavidir. Hastalığa sahip bireylerde güneş yanığı kolayca oluşabilir ve ciltlerinde
kırmızımsı çiller görünebilir ama cilt ve saç renkleri ailelerindeki diğer
bireylere göre her zaman daha açık kalır.
Genetik Değişiklikler/Etken Faktörler
Tietz sendromuna MITF genindeki mutasyonlar sebep
olmaktadır. Bu gen, bazı hücrelerin gelişiminde ve fonksiyonunda rol alan
proteinin yapımı için yardım sağlar. MITF proteini
melanositlerin (pigment üreten hücreler) gelişiminin kontrolüne yardım eder. Bu
melanositlerin içinde, bu protein saç, göz ve cilt rengini sağlayan melanin
pigmentinin üretimini sağlar. Melanositler ayrıca iç kulakta bulunur ve
işitmede önemli bir rol oynarlar. MITF proteini, rethina pigment epithel
dokunun gelişiminde de rol oynar. Dimerlerin (iki küçük alt birime sahip molekül) çoğu DNA’ya bağlanamaz ve bu da
melanositlerinin gelişimini ve melanin üretimini etkiler. İç kulaktaki melanositlerin azalması işitme
kaybına sebep olur. Azalan melanin üretimi, açık renk cilde ve saça sebep olur.
Belirti ve Semptomlar
Belirti
ve semptomlar genelde doğumda mevcut olabilir. Bunlar: şiddetli, iki kulakta da
sensörinöral işitme kaybı, açık tenlilik, açık renk saç rengi ve mavi gözler.
Hastaların yüzde 80-99’ununda bu belirtiler görülmektedir: gözde yapısal anomali, sağırlık, saç ve ten
renginin pigmentasyonun azalması ve beyaz kaşlara sahip olma.
Genetik Görülme Sıklığı
Tietz sendromu nadir görülen bir hastalıktır
ve görülme sıklığı kesin olarak bilinmiyordur. Etkilenen sadece birkaç aile
tıbbi literatürde görülmüştür.
Kalıtım Paterni/Deseni
Bu sendrom otozomal dominant
olarak kalıtılmaktadır.
Teşhis Yöntemleri ve Tedaviler
Tedavinin amacı, işitme kaybını
iyileştirmektir ve koklear implantasyonu düşünülebilir. Hastalığın teşhisi için
genetik test gerekmektedir (bu kaynaktan detaylı bilgi alınabilir: https://www.ncbi.nlm.nih.gov/gtr/conditions/C0391816/ ).
Hastalıkla İlişkili Genler
MITF genindeki mutasyonlar bu
hastalığa sebep olur.
Psödokolinesteraz eksikliği, kolin esterleri adı verilen genel anestezi
sırasında kullanılan bazı kas gevşetici ilaçlara duyarlılığın artmasıyla
sonuçlanan bir durumdur. Succinylcholine ve mivacurium gibi bu hızlı etkili
ilaçlar, nefes almada kullanılan kaslar dahil, hareket için kullanılan kasları
(iskelet kasları) gevşetmek için verilir. İlaçlar genellikle kısa cerrahi
prosedürler için veya bir solunum tüpünün hızlıca yerleştirilmesi gerektiğinde
acil durumlarda kullanılır. Normalde bu ilaçlar vücuttan birkaç dakika içinde
vücut tarafından parçalanır (bu sırada kaslar tekrar hareket edebilir). Bununla
birlikte, psödokolinesteraz eksikliği olan kişiler, ilaçlar verildikten birkaç
saat sonra kendi başlarına hareket edemeyebilir veya nefes alamayabilir.
Etkilenen bireyler, ilaçlar vücuttan temizlenene kadar nefes almalarına
(mekanik havalandırma) yardımcı olacak bir makine ile desteklenmelidir.
Psödokolinesteraz eksikliği olan kişilerde ayrıca lokal anestezik prokain
ve belirli tarımsal böcek ilaçları dahil olmak üzere diğer bazı ilaçlara
duyarlılığı artmış olabilir. Durum başka belirti veya semptomlara neden olmaz
ve anormal bir ilaç reaksiyonu meydana gelinceye kadar genellikle keşfedilmez.
Genetik Değişiklikler/ Etken Faktörler
Psödokolinesteraz eksikliğine BCHE genindeki mutasyonlar
neden olabilir . Bu gen, karaciğer tarafından üretilen ve kanda
dolaşan butirilkolinesteraz olarak da bilinen psödokolinesteraz enzimini yapmak
için talimatlar sağlar. Psödokolinesteraz enzimi, kolin ester ilaçlarının
parçalanmasında rol oynar. Enzimin vücutta başka işlevleri de vardır,
ancak bu işlevler iyi anlaşılmamıştır. Çalışmalar, enzimin sinir
sinyallerinin iletilmesinde rol oynayabileceğini göstermektedir.
Psödokolinesteraz eksikliğine neden
olan bazı BCHE gen mutasyonları, düzgün çalışmayan anormal
psödokolinesteraz enzimi ile sonuçlanır. Diğer mutasyonlar, psödokolinesteraz
enziminin üretimini önler. Fonksiyonel psödokolinesteraz enzim eksikliği,
vücudun kolin ester ilaçlarını etkin bir şekilde parçalama yeteneğini
zayıflatır ve anormal olarak uzun süreli ilaç etkilerine yol açar.
Psödokolinesteraz eksikliğinin genetik olmayan nedenleri de
olabilir. Bu durumlarda, durum kazanılmış psödokolinesteraz
eksikliği denir ; miras alınmaz ve bir sonraki nesle
aktarılamaz. Psödokolinesteraz enziminin aktivitesi böbrek veya karaciğer hastalığı, yetersiz beslenme, büyük yanıklar, kanser
veya bazı ilaçlar tarafından bozulmuş olabilir.
Belirti ve Semptomlar
PD genellikle kalıtsaldır. Çoğu durumda, anesteziye maruz kalana kadar rahatsızlık belirtileri veya semptomları yoktur.
Genetik Görülme Sıklığı
Psödokolinesteraz eksikliği 3.200- 5.000’de 1 kişide görülür. Pers
Yahudi cemaati ve Alaska yerlileri gibi bazı topluluklarda daha yaygındır.
Amerika Birleşik Devletleri’ndeki her 1,500 ila 2,500 kişiden yaklaşık birinde
görülür. Kafkasyalı Amerikalılar arasında erkeklerin neredeyse kadınlardan
iki kat daha fazla etkilediği görülüyor.
Kalıtım Paterni/ Deseni
Genetik nedenlerden dolayı, bu durum otozomal resesif paternde kalıtsaldırbu, her hücrede genin her iki kopyasında da mutasyon olduğu anlamına gelir. Çoğu zaman, otozomal resesif bozukluğu olan bir bireyin ebeveynleri, her hücrede değiştirilmiş genin bir kopyasına sahiptir ve taşıyıcı olarak adlandırılır. Gen mutasyonunu çocuklarına aktarabilirler, ancak genellikle hastalığın belirti ve bulgularını yaşamazlar. Bazı durumlarda, BCHE gen mutasyonlarının taşıyıcıları, kolin ester ilaçlarını vücuttan temizlemek için normalden daha uzun sürmektedir , ancak her hücrede değiştirilmiş genin iki kopyaları olduğu sürece. Teşhis Yöntemleri ve Tedavileri
PD’niz varsa, durumunuzu tanımlayan tıbbi bir bileklik takmak
isteyebilirsiniz. Aile üyelerine ameliyattan önce de test edilmelerini
söyleyin.
PD için tedavi olmasa da, tedaviler mevcuttur. Ameliyat sırasında
solunum durursa, solunum desteği sağlanabilir. Çoğu durumda, iyileşme
tıbbi yardıma ihtiyaç duymadan gerçekleşir.
Akut eozinofilik pnömoni(AEP)
akciğerlerde eozinofillerin ani birikmesiyle(pulmoner eozinofili) karakterize
nadir hastalıktır.Eozinofiller beyaz kan hücrelerinin bir tipidir ve bağışıklık
sisteminin bir parçasıdır.Genellikle allerjenlere,inflamasyonlara yada
enfeksiyona(özellikle parazitik olanlar) cevap olarak üretilirler ve özellikle
solunum bölgesinde aktiftirler.IAEP her yaşta meydana gelebilir ama yaygın olarak 20 ile 40 yaş arasındaki
sağlıklı bireyleri etkiler.AEP ile ilişkili yaygın semptomlar ani başlangıçlı
nefes darlığı(dispne) ve akut solunumda zayıflık,öksürük,yorgunluk,gece
terlemeleri,ateş ve istemeden kilo kaybı muhtemeldir.Bu hastalığın kesin sebebi
birçok hastada bilinmiyor(idiyopatik),yine de, son zamanlarda tütün kullanım
alışkanlıklarında değişiklikler ve ilaç alımı hastalığı tetikleyebilir.Kortikosteroidlerle
nüksetme yoktur,sonuç olumludur.
Giriş:
AEP’nin varlığı medikal literatürde
belirgin olarak 1989 da tanımlandı.AEP eozinofilik akciğer
hastalığının,intersiyel akciğer hastalıklarının geniş bit grubu,bir formu
olarak sınıflandırıldı.AEP Kronik Eozinofilik Pnömoniden farklıdır(CEP), Kronik
Eozinofilik Pnömoni yavaş ilerleyişle,akut solunum zayıflığının yok olmaya
doğru gidişiyle,sıklıkla tekrarlamayla ve sıklıkla astımla
ilişkilendirilmesiyle belirlenir.
İşaret ve Semptomlar:
AEP genellikle 1-7 günde aniden,hızlı
başlangıçlı semptomlarla karakterizedir.Yine de,bazı olgularda,semptomlar bir
aydan fazla daha yavaş bir şekilde gelişebilir.AEP sıklıkla gençlerde aksi
takdirde sağlıklı bireylerde gelişir.İlişkilendirilmiş semptomlar spesifik
değildir ve ateş,öksürük,solunumda zorluk(dispne) ve göğüs ağrısını
kapsarlar.Daha az yaygın olan semptomlar yorgunluk,kas ağrısı(miyalji),eklem
ağrıları ve karın rahatsızlığı ve ağrısını kapsar.AEP’de akut solunum
yetmezliği hızlı bir şekilde ilerleyebilir.Akut solunum yetmezliği kandaki
oksijen seviyesi şiddetli olarak düştüğünde(hipoksemi) meydana gelir sonuç
olarak hayatı tehdit edici solunum komplikasyonlarını ortaya çıkarma
potansiyeli vardır.Bu AEP’li bireylerde birkaç gün içinde hatta birkaç saat
içinde bile ortaya çıkabilir.Yaklaşık olarak hastaların üçte ikisinin mekanik
ventilasyona ihtiyacı olabilir.
AEP son derece nadir bir hastalıktır
ve özellikle bronkoalveolar lavajda
hücre sayımında değer farkının yokluğuyla infeksiyöz pnömoni ile karıştırılabilir.Araştırmacılar
hastalığın klinik anlamada daha iyi ve daha etkili bir şekilde tanımlanması
gerektiğini belirtmişlerdir.Örneğin bazı
araştırmacılar AEP nin ılımlı olan formunun olmadığına inanıyorlar fakat
bazı vakalarda spontan olarak gelişebilir.Bu ılımlı vakalarda hastalık şiddeti
az olan semptomlara ve komplikasyonlara sebeb olur.
Sebebleri:
IAEP’nin sebebi
bilinmiyor(idiyopatik).Araştırmacılar AEP’nin tanımlanamayan,spesifik olmayan
tetikleyici ajan nedeniyle vücutta eozinofil üretimine sebeb olan ve onların
akciğerlere yönlendirmesiyle geliştiğine inanıyorlar.Eozinofillerin aşırı
üretimi ve birikmesinin kesin sebebi bilinmiyor.
Mesleki faktörleride kapsayan birçok çevresel faktör toz ve dumana maruz kalmayı da AEP’yi tetikleyen
faktörler arasında göstermiştir.Tek bir faktör AEP’ye sebep olmamaktadır.Çoğunlukla
hastalığın gelişmesi için tetikleyici durumlara eğilimli hastalarda birçok
faktörün birarada bulunması gerekir.AEP’de tetikleyici faktör kişiden kişiye farklılık gösterebilir.
Çoğu vakada, özellikle hastalığın başlangıcından 3 ay
önce sigara içmeye başlamış bireylerde,geçici bırakmadan sonra içmeye devam
etmiş yada son zamanlarda günlük sigara kullanım sayısının artmasıyla birlikte
sigara kullanımının bu hastalığın gelişiminde anahtar rol oynadığına
inanılıyor.Medikal literatürdeki birçok rapor sigara kullanımının ile AEP
arasındaki ilişki etkilenmiş hastalarda altküme şeklinde sunulmuştur.Sigara
kullanımının AEP’nin gelişimi için kesin rolü bu gibi vakalarda tamamen
anlaşılamamıştır.
Mesleki faktörler çeşitli ve değişken tetikleyiciler
olarak gösterilmiştir;genellikle bunların önemli ortak özelliği solunan tozlara
maruz kalmaktır. Bu vakalarda hava yoluyla bulaşmak yada solunan ajanlar akciğerlere
zarar vererek AEP’nin tetiklenmesine sebep olmaktadır.
Bazı araştırmacılar için sitokinler(diğer bağışıklık
sistemi hücrelerinin stimüle olmasını yada inhibe olmasını sağlayan bağışıklık
sisteminin belirli hücrelerinden salgılanan özelleşmiş protein) eozinofilik
hastalıkların gelişmesinde rol oynayabilir.İnterlökin-5(IL-5) eozinofillerin
fonksiyonlarını ve gelişmelerini düzenleyici bir sitokindir.IL-5 ayrıca
akciğerlerde ve kan dolaşımında birikme sonucu oluşan eozinofillerin normal
parçalanmalarını(apoptoz) baskılar.AEP’nin tetikleyici ajanlarının mekanizması
ve kesin rolü için uygun durumda daha çok araştırma gereklidir.
Etkilenmiş Popülasyonlar:
AEP yaklaşık olarak erkekleri kızlara
göre 2 kat daha fazla etkiler.200 den az vaka medikal literatüre kaydedilmiştir
ve kesin prevelansı bilinmiyor.AEP bireyleri her yaşta etkileyebilir ama
genellikle en sık 20 ile 40 yaş arası bireylerde meydana gelir.
Teşhis:
AEP’nin teşhisi karakteristik
semptomların tanımlanmasına,detaylı hasta öyküsüne,klinik değerlendirmeye ve
özellikle bronkoalveoler lavaj(BAL) testi gibi çeşitli özel testlere dayanarak
konur.Parazitik enfeksiyonlar gibi pulmoner eozinofil sebepleri yada belirli
ilaçlara maruz kalma sistematik olarak sorgulanmalı.
Klinik Testleme ve Çalışma;
BAL olarak bilinen test,AEP’nin
anahtar teşhisidir.BAL sırasında,dar tüp(esnek bronkoskop) soluk borusundan
akciğerlerin içine doğru kaydırılır ve steril solüsyon hücrelerin içini
yıkayarak tüpten geçer(lavaj).Bu sıvı aspirasyonla birikmiştir ve sonra tüp
çıkartıldığında hücrelerin çalışmasına izin verir.AEP’li bireylerin BAL
sıvısında eozinofil seviyesi anormal bir şekilde yüksek çıkar(%25’ten büyük).Fiberoptik
bronkoskopi lokal anestezi altında yapılır;mekanik ventilasyonlu hastalarda
intratrakeal tüp vasıtasıyla yapılır.Arteryel kan gazları hipoksemiyi sıklıkla
şiddetli gösterir,birleştirilmiş akciğerde bu manevra sağdan sola doğru
şeklinde yansır.
Spesifik görüntüleme teknikleri göğüs
X-ray’ini de kapsayarak AEP’nin teşhisinin konulmasında yardımcı olarak
kullanılabilir,yinede anormallikler spesifik değildir.AEP’li kişilerin göğüs
X-ray’lerinde genellikle akciğerlerde beyaz çizgiler yada puslu yamalar
görülür.Göğüs CT’si ılımlı yada şiddetli seviyede bilateral plevral efüzyonla
ilişkili bilateral alveoler birleşmeyi gösterir ve interlobüler septal
kalınlaşma vardır ki hastalık için anlamlıdır.
Akut fazda pulmoner fonksiyon testleri tipik olarak kısıtlı paternleri gösterirler.
Tedavi:
AEP’li bireyler iki hafta için
reçetelendirilen yüksek dozlu kortikosteroidlere günler içerisinde cevap
verirler.Kortikostroid terapisinin sadece enfeksiyon sebebi pulmoner eozinofili
sonrası başlatılması göz ardı edilmelidir.Medikal literatürde,kortikosteroid
terapisinin dozu ve durasyonu büyük değişkenlik gösterir son zamanlarda yapılan
çalışmalar 2 haftalık tedavinin yeterli olabileceğini önermektedir.AEP’li
bireylerde kortikosteroid tedavisinin standardize dozu yoktur.Medikal
literatüre rapor edilen bireylerde başlangıçta ağız uygulamasından sonrasını
takiben damar yolundan kortikosteroid alınmıştır.Bazı vakalarda AEP hiçbir
tedavi olmadan iyileşir(spontan remisyon).Steroid terapisi durdurulduktan sonra
nüksetme olmamıştır.Uzun dönem prognozu mükemmeldir.
Sıklıkla hastalığın hızla gelişimi
yüzünden,birçok bireyin solunum desteği almak için yoğun bakım ünitesine kabule
gereksinimi vardır.Solunum desteği invaziv yada noninvaziv mekanik
ventilasyondan meydana gelmektedir.İnvaziv ventilasyon intratrakeal tüp
vasıtasıyla solunum desteğini sağlar.Noninvaziv ventilasyon hızlı gelişim
meydana gelene kadar kotikosteroidlerle yeterli olur sonrasında ventilatörle ve nazal yada yüz
maskesiyle solunum desteği sağlanır ve
mekanik ventilasyondan ayrılma imkanlı hale gelir(genellikle 1 haftadan daha az
sürede)




{kind=link}
{kind=link}