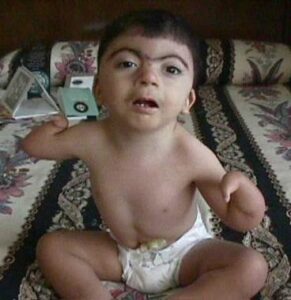
Oral-facial-digital sendromu, bir grup koşulun bir araya gelip ağız boşluğu (çene ve diş), yüz ve parmakların (el ve ayak) gelişimine etki etmesiyle oluşur. Bu koşullar birbirleriyle ilişki içerisindedirler.
Araştırmacılar OFD’nin en az 13 olası türünü tanımladılar. Bu farklı türler, belirti ve bulguların benzerliklerine göre sınıflandırıldı. Ancak, çeşitli türlerin özellikleri birbirleriyle büyük ölçüde çakıştı ve bazı türler de yeteri kadar iyi tanımlanamadı. OFD’nin sınıflandırma sistemi, araştırmacılar sendromdan etkilenen birey buldukça ve bu bozukluk hakkında daha fazla şey öğrendikçe gelişmeye devam etmektedir.
OFD’nin belirti ve bulguları oldukça kapsamlı, ancak, bu bozukluk türlerinin çoğu ağız boşluğu, yüz ve parmaklardaki gelişim problemlerini içermekte, birçok türü de beyin anormallikleri ve zihinsel engellik derecesiyle bağlantılı.
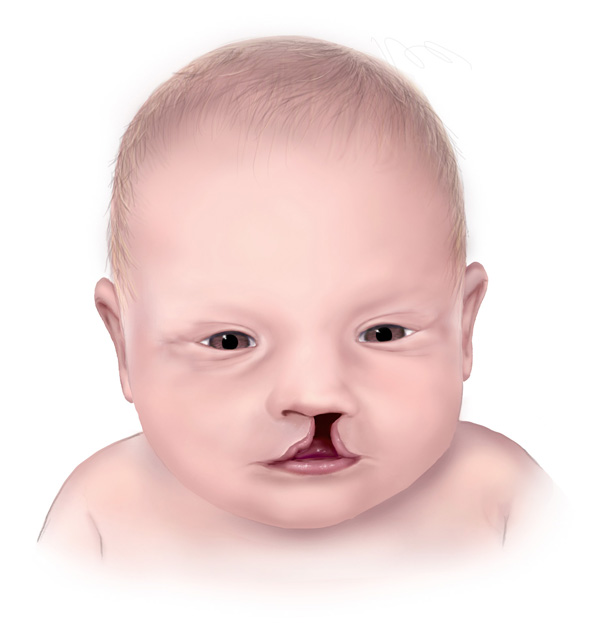
Oral-facial-digital sendromunun bir çok türü ağız boşluğu anomalilerini meydana getirir. Bunlardan bazıları; dilde yarık(split), olağandışı loblu dil(unusual lobed) ve kanser dışı tümör/yumruların dil üzerindeki gelişimidir. Ayrıca sendromdan etkilenen bireylerin ağzında fazla, eksik ya da bozuk dişler mevcut olabilir. Bir başka yaygın özellik ise ağzın üst kısmının açılmasıdır(yarık damak). OFD sendromuna sahip bazı insanlar ekstra doku bantlarına sahiptirler(hiperplastik frenula), bu anomali dudağın diş etine yapışması durumudur.
Kendine özgü yüzsel özellikler genellikle yarık dudak, geniş burun ve geniş aralıklı gözler(hypertelorism) içeren OFD ile ilişkilidir.
Parmaklardaki anormallikler OFD sendromuna sahip insanların hem el hem ayak parmaklarını etkileyebilir. Bu anormallikler bazı parmakların birleşmesi(syndactyly), parmakların genelden daha kısa olması(brachydactyly) ya da olağandışı kavisli olmasıdır(clinodactyly). Ayrıca fazla parmak olması da (polydactyly) OFD’nin çoğu türünde görülen bir özelliktir.
Diğer özellikler OFD sendromunun bir ya da birkaç türünde meydana gelir. Bu özellikler bozukluğun farklı türlerini ayırt etmede yardımcı olurlar. Örneğin, OFD sendromunun en bilinen türü OFD-I polikistik böbrek hastalığıyla ilişkilidir. Bu böbrek hastalığı sıvı dolu keselerin(kistlerin) büyümesiyle nitelendirilir. Keseler, böbreklerin kandan atık filtreme yeteneklerine müdahalede bulunurlar. OFD sendromunun diğer türleri de nörolojik problemler, beyin yapısındaki özel değişiklikler, kemik anomalileri, görme kaybı ve kalp rahatsızlıkları olarak tanımlanır.
Görülme sıklığı
OFD sendromunun tahmini sıklığı 50.000 ile 250.000 yeni doğan içinde 1’dir. Tür-I görülen bozukluğun çoğunluğunu oluşturur. OFD sendromunun diğer türleri çok nadir görülür; çoğu sadece bir yada bir kaç ailede tanımlanmıştır.
Genetik değişmeler
Sadece 1 gen, OFD1, OFD sendromuyla ilişkilidir. Bu gendeki mutasyonlar Oral-Facial-Digital sendromu Tür-I’in sebebidir. Ayrıca OFD1 geni mutasyonları Tür-VII olarak tanımlanan bozukluğu sahip ailelerde görülebilir; ancak, araştırmacılar şu anda Tür-I ve Tür-VII’nin aynı olduğuna inanıyorlar.
OFD1 geni, işlevi tam olarak anlaşılamayan bir protein yapmak için talimatlar sağlar. Bu durum beyin, yüz, kollar, bacaklar ve böbrekler gibi vücudun önemli kısımlarının erken gelişiminde önemli bir rol oynar. OFD1 genindeki mutasyonlar, yeterli miktardaki fonksiyonel OFD1 proteininden yapılmış hücreleri önler ve bu yapıların normal gelişimine engel olur. OFD sendromu Tür-I’in kendine özgü özelliklerini etkileyen proteinin bu sıkıntıyı nasıl oluşturduğu hala netleştirilmemiştir.
Araştırmacılar OFD sendromunun diğer türlerine sebep olan genetik değişimleri aktif bir şekilde araştırmaya devam ediyorlar.
Figür 1 Görüntü Kaynağı:https://ghr.nlm.nih.gov/art/large/polydactyly.jpeg
McKusick-Kaufman sendromu, genitoüriner malformasyonlar, polidaktili(çok parmaklılık; figür1) ve daha nadiren konjenital kalp hastalıkları veya gastrointestinal malformasyonlarla karakterize; neonatal dönemde ortaya çıkan çok nadir, genetik gelişimsel bir hastalıktır.
Belirti ve Semptomlar
McKusick-Kaufman sendromu; el ve ayak, kalp ve üreme sisteminin gelişimini etkileyen bir durumdur. Ekstra el parmakları ve/veya ayak parmakları (polidaktili), kalp anomalileri ve genital anormallikler: Üç özelliğin bir kombinasyonu ile karakterizedir.
McKusick-Kaufman sendromlu kızların çoğu, pelviste büyük bir sıvı birikmesi olan “hidrometrokolpos”(figür 2) olarak adlandırılan bir genital anomaliyle doğarlar. Hidrometrokolpoz, vajinanın bir kısmının gelişmemesi (vajinal agenezis) veya doğumdan önceki vajina tıkanıklığından kaynaklanır. Bu tıkanıklık, vajina ve rahim içinde sıvının oluşmasına, bu organların gerilmesine ve sıvı dolu bir kütleye yol açmasına sebep olur.
Figür 2 Görüntü Kaynağı: http://37.247.97.7/millipediatri/player.php?klasor_no=201703021431
McKusick-Kaufman sendromlu erkeklerde genital anormallikler, üretral açıklığın penis alt kısmında olması “hipospadias”(figür3), penisin aşağı doğru eğimli olmasın (chordee) ve inmemiş testis “kriptorşidizm”(figür4) şeklinde olabilir.
Figür 3 Görüntü Kaynağı: http://37.247.97.7/millipediatri/player.phpklasor_no=201703021431Figür 4 Görüntü Kaynağı: http://www.uroonkoloji.org/pdf/sunumlar/tarkan_soygur.pdf
McKusick-Kaufman sendromunun belirti ve bulguları, Bardet-Biedl sendromu olan başka bir genetik bozukluk olanlarla karıştırılmaktadır. Bardet-Biedl sendromu, McKusick-Kaufman sendromunda görülmeyen, kendine has çeşitli özelliklere sahiptir. Bunlar görme kaybı, gecikmiş gelişim, obezite ve böbrek yetmezliğini şeklindedir. Bu bulguların bir kısmı doğumda belirgin olmadığından, bebeklik ve erken çocukluk döneminde iki durumun ayırt edilmesi zor olabilir.
Görülme Sıklığı
Görülme sıklığı Eski Amish nüfusunda tanımlanmıştır, ki bu tahmin 10,000 kişiden 1’ini etkilemektedir. Amish olmayan populasyonlarda McKusick-Kaufman sendromunun görülme sıklığı bilinmemektedir.
Hastalığın Nedenleri
MKKS genindeki mutasyonlar McKusick-Kaufman sendromuna neden olur. Bu gen, ekstremitelerin, kalbin ve üreme sisteminin oluşumunda önemli bir rol oynayan bir proteinin üretilmesi için talimat sağlar. Protein yapısı, diğer proteinlerin katlanmasına yardımcı olan bir protein türü olan bir “şaperonine”le benzerdir. Proteinler vücutta normal işlevlerini yerine getirmek için doğru 3 boyutlu şekle katlanmalıdır. MKKS proteininin yapısı bir şaperonine benzese de, bazı yeni çalışmalar protein katlanmasda bu proteinin birincil işlevi olmadığını düşündürmektedir. Araştırmacılar, MKKS proteininin hücre içindeki diğer proteinlerin taşınmasında da rol oynayabileceğini düşünüyorlar.
McKusick-Kaufman sendromunun altında yatan mutasyonlar MKKS proteininin yapısını değiştirir. Değişmiş protein, doğumdan önce vücudun çeşitli bölümlerinin gelişimini bozsa da, MKKS mutasyonlarının bu bozukluğun spesifik özelliklerine nasıl yol açtığı belirsizdir.
Kalıtım Şekilleri
Kalıtım otozomal resesiftir. Bu da her bir hücredeki genin kopyalarının mutasyona sahip olduğu anlamına gelir. Otozomal resesif bir rahatsızlığı olan bir bireyin anne-babalarının her biri mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Teşhis Yöntemleri
.Radyolojik Görüntüleme Yöntemleri
.Genetik Testler
.Gen Haritası Çıkarma
Görüntü Kaynağı: http://www.eurorad.org/eurorad/case.php?id=12659
Tedavi
Tedavisi daha anomalilere yönelik şekilde yapılmaktadır. Duruma göre gerek cerrahi gerek medikal tedavi yapılmaktadır. Nokta atışı tedaviler üzerine hücresel düzeyde çalışılıyor.
Hastalığın Diğer İsimleri: Ektopik nörofizyoloji, PSIS
Genel Bilgi
PSIS doğuştan meydana gelen bir hipofiz sapı rahatsızlığıdır. Çok ince veya kesintiye uğramış hipofiz sapı, hipofizin bulunduğu yerin normalden farklı olması veya ön (posterior) hipofizin hiç olmaması, arka (anterior) hipofizin normalden küçük olması ya da olmaması gibi sebeplerle kalıcı olarak büyüme hormonu bozukluklarına yol açmaktadır.
Yeni doğanlardaki işaret ve semptomları : düşük kan şekeri (hypoglycemia), sarılık, üreme organlarında (testis, penis,skrotal kese) doğuştan görülen anormallikler gibi. Çocukluktan sonra kısa boy, nöbetler, düşük damar basıncı (düşük arteriyel basınç, hipotansiyon) ve / veya zihinsel gecikme de görülebilir. Sadece bazı hastalara özgü olarak; sebebi bilinmemekle birlikte görme bozuklukları (septooptik displazi) ve kemik iliği yetmezliği (Fanconi anemia) de semptomlar arasında yer alabilir. HESX1, LHX4, OTX2, SOX3, ve PROKR2 isimli genlerde nadir olarak görülen mutasyonlar da ırsi sebeple hasta olan kişilerde görülebilir. Aynı zamanda MRI görüntüleri ile gözlemlenen, hastalığa özgü karakteristik bulgularla teşhis ve tanı süreci sağlamlaştırılır. Tedavi ise, hormon tedavisi ve doğumdan itibaren müdahaleye başlanarak özellikle hormon ve hormon dengesi bozukluğunun ve zihinsel geriliğin engellenmesi ile mümkündür[1]
Sonuç olarak PSIS, hipofiz yetmezliğinin çok da nadir olmayan bir sebebini oluşturmaktadır. Ancak yeteri kadar tanınmadığından genellikle hastalar idiyopatik BH eksikliği veya hipofiz yetmezliği tanısı almakta çoğu zaman da tanı gecikmesine neden olmaktadır. Bu tür olgularda PSIS tanısının göz önünde bulundurulması ile özellikle çoklu hormon eksikliğinin etyopatogenezi aydınlatılabilir. Böylece ailelerin doğru bilgilendirilmesi ve hasta takiplerinin daha doğru yapılması sağlanabilir[2]
Belirti ve Semptomlar
Aşağıdaki tabloda hastaların sahip olabileceği olası semptomlar gösterilmektedir. Çoğu hastalıkta olduğu gibi bunda da semptomlar (belirtiler) hastaya göre çeşitlilik göstermektedir.[3](Tablo 2)
Hastalığın tam sebebi henüz açıklanamamıştır. HESX1 (3p21.2-p21.1) geninin transkripsiyon faktörlerindeki mutasyon hem PSIS hastalarında hem PSIS bağlantılı görme bozukluklarında (septooptic dysplasia) gözlemlendi. LHX4 (1q25) genindeki mutasyon da PSIS’ye yol açmaktadır. Bunlarla beraber, olguların çoğunda herhangi bir genetik neden bulunmamakla birlikte, ailesel sebeplerin varlığı ve PSIS’nin özellikle gözlerin konjenital anomalileri ve mikrofallus durumu ile birlikteliği, bir antenatal (doğum öncesi) kaynaklı olduğunu düşündürmektedir.[4]
Ektopik nörohipofiz, infundibulumda (hipofiz sapı) distal (uç taraf) olarak yer değiştiren bir arka hipofiz bezi anomalsidir.• Tanı, genellikle sagital düzlemde en iyi görülen parlak T1 ağırlıklı (T1-weighted) bir görüntü gösteren manyetik rezonans görüntüleme ile yapılır.• Ektopik nörohipofizin, sadece MRI ile tesadüfi bir bulgu olduğu düşünülebilir ancak periventriküler nodüler heterotopia, perisilvian sendromu, holoprozensefali ve septo-optik hipofiz displazisi gibi çeşitli serebral malformasyonlarla (beyin ile ilgili bozukluklar) ilişkilidir.• Klinik gözlem, çoğunlukla diyabet insipidustan ziyade büyüme hormonu eksikliği (adenohipofizyal kaynaklı) veya panhipopitüitarizm ile ilişkili endokrinolojiktir.• Bazı durumlarda renal anomaliler (böbrek ile ilgili bozukluklar) ile ilişkili olabilir.
• Köken, erken fetal yaşamda (anne karnında), yani genetik olarak belirlenmiş bir konjenital anomalide gelişir.
• Çoklu genetik mutasyonlar (genetik bozunum) veya delesyonlar (gen silinmesi) ve nadiren kromozomatiler (kromozom bozuklukları) ektopik nörohipofiz ile ilişkili olarak tanımlanır, bu nedenle bu durum bir sendrom olarak kabul edilmelidir.
Genetik Görülme Sıklığı
Hipofiz sapı kesilme sendromu (PSIS), tahmini insidansı (görülme sıklığı) 0.5 / 1.000.000 olan nadir bir hastalıktır.[6]
Çeşitli formlarından; otozomal baskın, otozomal çekinik, kalıtsal değil.[8]
Teşhis Yöntemleri ve Tedaviler
Klinik bulgulardan ve stimülasyon testinden yola çıkarak konulan teşhislerde düşük büyüme hormonu (GH) pikine ve düşük insülin bağlı büyüme faktörlerine dayanarak hastalıktan şüphelenilebilir. Kalıcı GH bozuklukları ise kesin tanı konulmasında etkendir. GH bozukluklarının bağımsız mı yoksa arka hipofiz bezi bozuklukları ile bağlantılı mı olduğunu (PSIS hastalarında %70 olarak görülen durumdur) test etmek gereklidir. MRI ölçümlerinden PSIS tanısı konulduğu takdirde başka olası tanı yoktur.[9]
Tedavi eksik hormonların; özellikle GH, tiroksin, hidrokortizon ve puberte, seks steroidlerinin yenilenmesine dayanır. Hipoglisemi (düşük kan şekeri) ve ikincil adrenal yetmezliği ve bunlarla ilişkili serebral (beyinle ilgili) ve vital (hayati) riskleri önlemek için, doğumda PSIS tanısı koymak önemlidir. Zamanında teşhis ve tedavi ile prognozu iyidir.
Tanı ve tedavide gecikme, hipoglisemi ve / veya kortizol eksikliğine bağlı hipotansiyon ve / veya tiroid eksikliğine bağlı zihinsel gerilikten dolayı nöbetlere neden olabilir. Bu risklerin bir sonucu olarak, PSIS hastalarında mortalite (ölüm) ve morbidite, genel olarak 2 yaşından önce hastalığı ortaya çıkan hastalarda daha yüksektir.[10]
Şekil 1. Manyetik rezonans görüntülemede hipofiz sapı kesilme sendromunun tipik özellikleri. (A) Ektopik arka hipofiz ile birlikte ince hipofiz bezi (27). (B) Hipofiz sapının yokluğu (7,28). (C) İnce hipofiz sapı (29,34). Detaylı resim için : https://www.researchgate.net/publication/266086657_Pituitary_stalk_interruption_syndrome_Case_report_of_three_cases_with_review_of_literaturKAYNAK: orphanet
Doğumsal Anozmi, doğuştan koku alma duyusunun tam ya da kısmi olarak kaybedilmesi durumudur. İzole bir anormali (ek semptom yok) olabileceği gibi spesifik bir genetik bozuklukla (Kallmann sendromu veya Doğumsal Ağrı Duymazlık gibi) ilişkili olabilir. Bazı ailevi vakalar bildirilmiş olmasına rağmen izole doğumsal anozmi genellikle sporadiktir[1]. Çoğu zaman İzole Doğumsal Anozmi’nin genetik nedeni bilinmemekle beraber koku almamızı sağlayan sistemin (olfaktör sistem) gelişimi sırasında oluşan hataların neden olduğu ve bunun 18. kromozomdaki bir genin mutasyonundan kaynaklanıyor olabileceği düşünülmektedir. Ne yazık ki, bu hastalığın tedavisi yoktur.
Genetik Değişiklikler/Etken Faktörler
Doğumsal Anozmi, izole bir anormali (ek semptom yok) olabileceği gibi spesifik bir genetik bozuklukla (Kallmann sendromu veya Doğumsal Ağrı Duymazlık gibi) ilişkili de olabilir.
İzole doğumsal anozmi (ek semptom yok) çoğu hastada aile öyküsü olmaksızın sporadik olarak ortaya çıkmaktadır. Bu durumun temel nedeni bilinmemektedir. Bilim insanları bu durumun anne karnında olfaktör sistemin (koku almamızı sağlayan sistem) anormal gelişmesinden kaynaklanabileceğinden şüphelenmektedir. Bu anormal gelişime, burundan beyne bilgi taşıyan yoldaki bozulmalar ya da kokuyu işleyen beyin bölümünün malformasyonları[2] gibi burun boşluğu anormalileri dahil edilebilir.
İzole doğumsal anozminin birden fazla aile üyesini etkilemesi durumunda, genetik bir neden akla gelmektedir. Yapılan bir çalışmada, izole doğumsal anozmiden etkilenen bazı kişilerin PROKR2 veya PROK2 geninde değişikliklere (mutasyonlara) sahip olduğu bildirilmiştir. Bu genler daha önce Kallmann sendromu (Doğumsal Anozmi ve diğer semptomlarla ilişkili kalıtsal bir sendrom) olan hastalarda da bildirilmiştir. Yapılan bir başka çalışmada, Anozmiden muzdarip iki erkek kardeşin CNGA2 geninde bir mutasyon olduğu bulunmuştur. Buna rağmen çoğu aile içi izole doğumsal anozmi olgusunda neden tam olarak bilinmemektedir.
Belirti ve Bulgular
Belirtiler aşağıdaki tabloda özetlenmiştir. Çoğu hastalıkta belirtiler kişiden kişiye değişiklik gösterebilmektedir yani aynı hastalığa sahip kişiler aynı belirtileri göstermek zorunda değildir. Tablonun kaynağı HPO (Human Phenotype Ontology) isimli bilgi bankasıdır. Dilerseniz HPO kodunu kullanarak belirtiler hakkında daha fazla bilgi sahibi olabilirsiniz (İngilizce).
Terimler
Diğer İsimler
Daha Fazla Bilgi için HPO kodu
Hastalığa sahip kişilerin belirtilere sahip olma yüzdeleri HPO bilgi bankasında gösterilmemiştir.
Yapılan çalışmalar, yaklaşık her 10.000 kişiden 1’inin doğumsal anozmiden muzdarip olduğunu göstermektedir. Ancak yapılan çalışmalar sadece izole doğumsal anozmi hastalarını içermemekte bunun yanında spesifik bir genetik bozukluğun neden olduğu (Kallmann sendromu veya Doğumsal Ağrı Duymazlık gibi) diğer doğumsal anozmileri de içermektedir. Orphanet nadir hastalıklar veritabanında sadece İzole Doğumsal Anozmi’nin görülme sıklığı 1.000.000 kişide 1’den az olarak belirtilmiştir.
Kalıtım Deseni (Paterni)
İzole doğumsal anozmi (ek semptom yok) çoğu hastada aile öyküsü olmaksızın sporadik olarak ortaya çıkmaktadır. Nadiren, birden fazla aile üyesi etkilenebilir. Bu ailelerde hastalığın kalıtımı azalmış penetransla birlikte otozomal dominant olarak görülmektedir. Otozomal dominant kalıtım, her hücredeki sorumlu genin sadece bir kopyasındaki değişikliğin (mutasyonun) hastalığa neden olmak için yeterli olduğu anlamına gelir. Azalmış penetrans, mutasyona uğrayan ya da kalıtımsal olarak mutasyona sahip olan herkesin etkilenmeyeceği anlamına gelir. Otozomal dominant kalıtılan bir mutasyona sahip olan birey çocuk sahibi olduğunda (etkilenmiş olsun olmasın), her bir çocuğun bu mutasyona sahip olma olasılığı % 50’dir. Azaltılmış penetransı olan durumlar için ise mutasyonu alan bir çocuğun hastalık özelliklerini gösterip göstermeyeceğini öngörmek mümkün değildir. İzole Doğumsal Anozmiden muzdarip pek çok ailesel vakada genetik neden bilinmemektedir.
Doğumsal Anozmi, Kallmann sendromu ve Doğumsal Ağrı Duymazlık gibi kalıtsal genetik bozukluklarla da ilişkili olabilir. Doğumsal Anozminin bu bozukluklarda kalıtımı ilişkili bozukluğun kalıtımı ile aynıdır. Örneğin, Kallmann sendromu, altta yatan genetik nedene bağlı olarak otozomal dominant, otozomal resesif veya X’e bağlı resesif bir şekilde kalıtılabilir (birkaç farklı gendeki mutasyonlar bu duruma neden olabilmekte). Doğumsal ağrı duymazlık, otozomal resesif kalıtıma sahiptir.
Teşhis Yöntemleri ve Tedaviler
İzole Doğumsal Anozmi (ek semptom yok) teşhisi diğer olasılıklar dışlanarak konur. Yani teşhis koyarken koku alma duyusunun kaybına yol açabilecek bilinen diğer tüm koşullar dışlanmalıdır. Hastalıktan muzdarip bir kişinin daha önce koku aldığına dair bir anısı olmaması durumunda, doğumsal anozmi teşhisini desteklemek için aşağıdaki testler yapılabilir:
Koku duyusuna etki edebilecek diğer koşulları aramak için kapsamlı fizik muayene ve tıbbı öykü sorgulaması
Koku Testleri
BT veya MRI gibi beyin görüntüleme teknikleri beynin koku duyusunu işleyen kısmında yer alan malformasyonları aramak için kullanılabilir.
Burun endoskopisi, burun boşluğundaki koku duyusunu etkileyebilecek anormallikleri saptamak için yapılabilir.
Olfaktör(Koku Siniri) Sinir Testi burundan beyne bilgi taşıyan yoldaki bozulmaları değerlendirmek için yapılabilir.
Ne yazık ki, bu hastalığın tedavisi yoktur.
Hastalıkla İlişkili Genler
Hastalıkla ilişkili olduğu düşünülen birkaç gen ortaya konmuştur. Bunlardan ilki Xq28 bölgesindeki CNGA2 geni iken bir diğeri Xq25 bölgesindeki TENM1 genidir. Ayrıca 18p11.23-q12.2 bölgesindeki ANIC geninin de hastalıkla ilişkili olabileceği gösterilmiştir. Yapılan bir başka çalışmada, izole doğumsal anozmiden etkilenen bazı kişilerin PROKR2 veya PROK2 geninde değişikliklere (mutasyonlara) sahip olduğu bildirilmiştir
Retinitis Pigmentosa(RP), kalıtımsal hastalıklar grubunda olup gözün ışığa duyarlı kısmındaki(retina) hücrelerin yıkımı ile seyreden nadir hastalıklardan biridir. RP retinada yer alan çubuk ve koni hücrelerinin ölümüne yol açar. Bu durum ilerleyici görme kaybına neden olur. Hastalığın ilk bulgusu genellikle gece körlüğüdür. İlerleyen evrelerde ise bireylerde Tünel Görüşüne(Periferal Görme kaybı) neden olur. Hastalığın son evresinde ise merkezi görme kaybı oluşur. RP gelişim etyolojisinde en az 50 gendeki herhangi bir mutasyon yer almaktadır. Hastalığın kalıtsal aktarımı Otozomal Dominant, Otozomal Resesif ya da X’e bağlı olabilir. Tedavi seçenekleri ilerleyici görme kaybını yavaşlatmak için yüksek ışıktan kaçınma, düşük görüş yardımı ve A vitamini desteğidir. Araştırmacılar gelecekte Gen terapisi, Kök hücre nakli ve Prostetik İmplant gibi yeni tedavi seçenekleri geliştirmek için çalışıyorlar.
Sıklık
Retinitis Pigmentosa Amerika ve Avrupa’da 3.500 ya da 4.000’de bir görülen bir hastalıktır.
Belirtiler
Aşağıdaki tablo bu hastalığa sahip insanlarda görülebilecek semptomları listeler. Çoğu hastalıkta ortaya çıkan semptomlar kişiden kişiye değişebilir. Yani Retinitis Pigmentosa’ya sahip tüm insanlar listede yer alan semptomların tümünü göstermeyebilir.
Medikal Terimler
Diğer İsimler
Anormal elektroretinogram
Anormal retinal pigmentasyon
Abnormal retinal vaskülarite
Anormal retinal kan damarları
Anormal testis
Burun kanatlarında anteversiyon
Atipik deri skarları
Atipik skarlar
Körlük
İletim tipi işitme kaybı
İletim tipi sağırlık
Hipogonadizm
Gonad aktivitesinde düşme
Penis Hipoplazisi
Gelişmemiş penis
Mental Retardasyon
Zeka geriliği
Nistagmus
Ritmik göz hareketleri
Optik atrofi
Fotofobi
Işığa aşırı hassasiyet
İlerleyici gece körlüğü
Sensörinöral işitme kaybı
Geniş burun köprüsü
Katarakt
Göz lensinin bulanıklaşması
Glokom
Hiperinsülinemi
Keratokonus
Kabartılı kornea
Obezite
Yüksek kiloya sahip olmak
Oftalmopleji
Göz kasları felci
Hiperrefleksi
Artmış refleksler
Tip 2 diyabet
İnsülin bağımlı olmayan diyabet
Periferal görme alanında darlık
Sınırlı periferal görüş
Niktalopi
Gece körlüğü
Çubuk-Koni Distrofisi
Bu Bilgi “Human Phenotype Ontology(HPO)” veritabanından alınmıştır. HPO tıbbi kaynaklarda tanımlanmış semptomlar üzerinde bilgi toplayan bir veritabanıdır. Düzenli olarak güncellenir. Daha fazla bilgi için https://hpo.jax.org/app/
Kalıtım
RP gelişim etyolojisinde en az 50 gendeki herhangi bir mutasyon yer almaktadır. RP ile ilişkili genler retinadaki fotoreseptörlerin yapım, yıkım ve fonksiyonlarında önemli rol oynarlar.
Retinada 2 çeşit fotoreseptör hücre bulunur. Bunlar çubuk ve koni hücreleridir. Koni hücreleri renkli görmeyi de içeren gün ışığı görüşünü sağlarken, çubuk hücreleri düşük ışıktaki görmeden sorumludur. Bu hücrelerdeki ileyleyici dejenerasyon insanlarda RP ile ortaya çıkan görme kaybının karakteristik yapılanmasına neden olur. Çubuklar tipik olarak konilerden önce bozulur. Bu yüzden ilk semptom genellikle gece körlüğüdür. Gündüz görüşü daha sonra çubuk ve koni hücrelerinin her ikisi bozulduğunda ortaya çıkar.
Hastalığın kalıtsal aktarımı Otozomal Dominant, Otozomal Resesif ya da X’e bağlı olabilir.
Bu genlerin 20’den fazlası hastalığın Otozomal Dominant formu ile ilişkilidir.
Bu genlerin 35’den fazlası hastalığın Otozomal Resesif formu ile ilişkilidir.
Bu genlerin 6’dan fazlası hastalığın X’e Bağlı formu ile ilişkilidir.
Otozomal Dominant(OD) Retinitis Pigmentosa
OD kalıtımda her bir hücredeki sorumlu genin tek bir kopyasındaki mutasyon hastalığın ortaya çıkması için yeterlidir. Rhodopsin genindeki mutasyonlar OD kalıtımın en sık(%20-30) sebebidir. Bazı vakalarda hasta kişi mutasyona uğramış geni yine hasta olan anne-babasından alır. Bazı vakalarda da aile öyküsü olmamasına rağmen mutasyon ilk kez bir bireyde ortaya çıkabilir. OD kalıtıma sahip bir hastanın doğan çocuklarının her birinin mutasyonlu gene sahip olma olasılığı %50’dir.
Otozomal Resesif(OR) Retinitis Pigmentosa
OR kalıtımda hastalığın ortaya çıkması için her bir hücredeki sorumlu genin her iki kopyasında mutasyon olması gerekir. USH2A genindeki mutasyonlar OD kalıtımın en sık(%10-15) sebebidir. Etkilenen insanlar sorumlu genin mutasyona uğramış kopyasını hasta ve/veya taşıyıcı olan anne-babasından alır. OR taşıyıcılar genellikle tipik olarak etkilenmezler/semptom göstermezler. OR kalıtıma sahip 2 taşıyıcının çocuklarının her biri:
%25 ihtimalle hasta doğar.
%50 ihtimalle anne-babası gibi etkilenmez ancak taşıyıcı olur.
%25 ihtimalle hiç etkilenmez ve taşıyıcı olmaz.
X’e Bağlı Aktarılan Retinitis Pigmentosa
RPGR ve PP2 genlerindeki mutasyonlar X’e bağlı kalıtımın en sık sebepleridir. Sorumlu gen X kromozomu üzerine yerleşmiştir. Kadınlar(XX) 2 X kromozomuna sahipken erkekler(XY) bir X ve bir Y kromozomuna sahiptir. X kromozomunda mutasyon taşıyan tüm erkekler etkilenecektir. Kadınlar ise etkilenebilir veya etkilenmeyebilir. Çünkü kadınlar genin normal kopyasına sahip başka bir X kromozomuna daha sahip olabilirler.
Hasta bir erkeğin bütün kız çocukları mutasyona uğramış geni alacaklardır, erkek çocukları ise etkilenmeyeceklerdir.
Hasta bir kadının erkek çocukları %50 ihtimalle etkilenecek ve hasta olacaklardır. Kız çocukları ise %50 ihtimalle etkilenecektir, hasta olacak veya hasta olmayıp taşıyıcı olacaklardır.
RP ile ilişkili genlerin bazıları başka hastalıklarla da ilişkilidir. Örneğin Çubuk-Koni Distrofisi(CRD). CRD ile RP benzer göz semptomlarına sahiptir. CRD’de RP’den farklı olarak önce koni hücreleri bozulur, daha sonra çubuk hücreleri bozulur. Böylece gün ışığı ve renkli görüş, gece görüşünden daha önce bozulur.
RP ile ilişkili diğer bazı genler: RHO, RPGR, RP2, PRPH2, RP7, RP9, RPE65 …vb
Teşhis
Nadir bir hastalığının tanısını koymak sıklıkla zor olabilir. Hekimler hastalığın teşhisini koyarken genellikle kişinin tıbbi geçmişine, semptomlarına, fizik muayenesine ve laboratuvar testlerine bakarlar. Aile hikayesi çoğu zaman yanıltıcı olabilir. Bu yüzden bazı durumlarda sorumlu gen Moleküler Genetik Testler ile belirlenmelidir.
Tedavi
Var olan tedavi seçenekleri ilerleyici görme kaybını yavaşlatmayı amaçlar ve bunun için yüksek ışıktan kaçınma, düşük görüş yardımını içerir. Bazı hekimler aynı zamanda A vitaminini bir tedavi seçeneği olarak tercih ederler. Fakat çok fazla A vitamini almak toksik etkiler yaratabilir ve A vitamininin tedaviye katkısını azaltabilir.
Çalışmalar, balıklarda doğal olarak bulunan ve omega-3 yağ asitlerinden olan Dokozahekzaenoik Asit(DHA) ile ilişkili olan potansiyel bir tedavi yöntemi olabileceğini gösteriyor. DHA’nın retinal hücrelerde yapısal bir rol oynadığı bilinmesine rağmen tedaviye katkısı olup olmadığının tam olarak belirlenebilmesi için daha fazla araştırmaya ihtiyaç duyulmaktadır.
Son zamanlardaki araştırmalar; gen terapisi, retinal transplantasyon ve retinal protez kullanımını içeren yeni tedavi yöntemlerine odaklanmış durumdadır. Kök hücre nakli, enjeksiyon ve kök hücrelerinin retina içine entegrasyonu ile sağlanıp bu hücrelerin ölü hücrelerle yer değiştirmesini ve görüş için gerekli olan enzimler ve kimyasalların sağlanmasını amaçlamaktadır.
Eğer hastalığa sebep olan mutasyonun lokalizasyonu biliniyorsa gen terapisi potansiyel olarak kullanılabilir. Bu tedavi kaybolan ya da anormal üretilen proteinlerin yeniden ve düzgün üretimini sağlar.
FDA Onaylı Tedaviler
Voretigene Neparvovec (Luxturna®)
FDA onaylı endikasyon: Adeno ilişkili bir virüs vektörü bazlı gen terapisi, biallelik RPE65 mutasyonu ile ilişkili retina distrofisi olan hastaların tedavisinde endikedir.
Juvenil miyoklonik epilepsi (JME), miyoklonik jerklerle (kolların veya bacakların hızlı, istem dışı kasılması), jeneralize tonik-klonik nöbetler ve bazen absans nöbetler ile karakterize bir epilepsi sendromudur. JME nöbetleri genellikle insanlar sabah ilk uyandığında ortaya çıkar. Nöbetler uyku eksikliği, aşırı yorgunluk, stres veya alkol tüketimi ile tetiklenebilir. Başlangıç genellikle normalde sağlıklı olan çocuklarda adolesan çağlarında ortaya çıkar. JME’nin nedenleri çok karmaşıktır ve tam olarak anlaşılamamıştır. GABRA1 ve EFHC1 genleri dahil birkaç genin birinde ortaya çıkan mutasyonlar, bu hastalığa neden olabilir veya duyarlılığı artırabilir. Hastaların antikonvülsanlarla ömür boyu tedavileri gerekse de, prognozları genellikle iyidir. (1)
Genetik Değişiklikler/Etken Faktörler
JME’nin kesin nedeni bilinmemektedir. Kafa travması, beyin tümörü veya ensefalit gibi durumlarla ilişkili değildir.
Aile öyküsü ve genetik faktörler JME riskinde güçlü bir rol oynamaktadır. Etkilenen insanların yaklaşık üçte birinin epileptik nöbetleri olan bir akrabası vardır ve birçok ailede spesifik genetik mutasyonlar bulunmuştur. GABRA1 ve EFHC1 genlerindeki ve henüz tanımlanmayan diğer genlerdeki mutasyonlar JME’ye neden olabilir veya duyarlılığı artırabilir.
GABRA1 geni, hücre zarını geçen klorür iyonlarının akması ile ilgili bir proteinin yapılması için talimatlar sağlar. Klorür iyonlarının akışı, hücre içinde sinir hücreleri (nöronlar) arasındaki sinyalleşmeyi engelleyen ve beynin çok fazla sinyalle aşırı yüklenmesini önleyen bir ortam oluşturur. GABRA1 genindeki mutasyonlar beyindeki nöronların aşırı uyarılmasına yol açar ve nöbetlerle ilişkili anormal beyin aktivitesini tetikler.
EFHC1 genindeki mutasyonlar az sayıda insanda JME ile ilişkilendirilmiştir. EFHC1 geni, nöron aktivitesinde de rol oynayan bir proteinin yapılmasına yönelik talimatlar sağlar, fonksiyonu tam olarak anlaşılmamış olmakla birlikte, aynı zamanda nöronların aşırı uyarılmasına ve nöbetlerin tetiklenmesine de yol açabilir.
JME, aşağıdaki belirtilerle ortaya çıkabilir:
Uyku eksikliği
Psikolojik stres
Alkol ve uyuşturucu kullanımı
İlaç uyumsuzluğu
Strobe ışıkları gibi titreyen ışıklar
Regl
Günün saati – genellikle sabahlar (1)
Belirti ve Semptomlar
JME’nin belirtileri ve semptomları şunlardır:
Kolların ve bacakların hızlı, istem dışı kasılması olarak tanımlanan ve hastalığın ayırt edici özelliği olan miyoklonik jerkler veya nöbetler vakaların yaklaşık % 17’sinde tek belirti olabilirler; Olguların yaklaşık % 20’sinde nöbetler kümeler halinde görülür, vücudun sadece bir tarafını (tek taraflı) etkiler ve tonik-klonik nöbet öncesi başlar.
Jeneralize tonik-klonik nöbetler, miyoklonik jerklerin başlamasından birkaç ay sonra ortaya çıkar.
Absans nöbetleri, genellikle 5 ve 16 yaşlarında ortaya çıkan ilk semptomdur.
Miyoklonik status epileptikus, JME’yi en çok ilgilendiren problem olarak kabul edilir. Çoklu miyoklonik nöbetler hemen durmadığında, uyku yoksunluğundan ya da kaçırılan nöbetlerden sonra bu durum görülür. (1)
Genetik Görülme Sıklığı
JME, idiyopatik jeneralize epilepsilerin yüzde 25 ile 30’unu ve tüm epilepsi vakalarının yüzde 10’unu oluşturur. Epilepsi riski yüzde 20 olan 20 yaş baz alındığında, JME insidansı 1000 ile 2000’de 1 olarak tahmin edilmektedir.
JME’deki cinsiyet oranının genel olarak eşit olduğu düşünülmektedir, ancak birkaç çalışma kadınlarda 2.9:1’e kadar olan bir üstünlüğü bildirmiştir. (2)
Kalıtım Paterni/Deseni
JME’nin kalıtım paterni tam olarak anlaşılamamıştır. Durum GABRA1 genindeki mutasyonlardan kaynaklandığında otozomal dominant bir kalıba kalıtılır, bu da her bir hücrede değiştirilmiş genin bir kopyasının bozukluğa neden olmak için yeterli olduğu anlamına gelir. EFHC1 genindeki mutasyonların neden olduğu juvenil miyoklonik epilepsinin kalıtım paterni bilinmemektedir.
JME epilepsisi ailelerde çalışabilse de, çoğu vakada aile öyküsü bulunmayan kişilerde görülür. (3)
Teşhis Yöntemleri ve Tedaviler
Genetik veya nadir bir hastalık için tanı koymak genellikle zordur. Sağlık uzmanları tanı koymak için genellikle hastanın anamnezine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına bakar.
Alkol kullanımı ve uyku yoksunluğu gibi durumlardan hastayı kaçındırmak faydalı olabilir. Bununla birlikte, anti-konvülsanlarla medikal tedavi genellikle gereklidir ve iyi tolere edilir. Hastaların çoğu tek bir ilaç, en yaygın olarak valproik asit ile iyi kontrol edilebilir. Ayrı ayrı veya kombinasyon halinde kullanılabilecek diğer ilaçlar arasında lamotrijin, levetirasetam, klonazepam ve topiramat bulunur. (1)
Hastalığın Seyri
Nöbetler genellikle nöbet ilaçları ile iyi kontrol edilir ve çalışmalar nöbetlerin yaşamın dördüncü on yılından sonra düzelme eğiliminde olduğunu göstermektedir. Bir kişi nöbet geçirmiyor olsa bile, nöbet ilacı genellikle yüksek tekrarlama riski nedeniyle devam eder (özellikle daha şiddetli formda olanlarda). İnsanların çoğunluğu için ömür boyu tedavi gereklidir. (1)
Nonsendromik holoprozensefali, kafa ve yüz yapısını da etkileyen beyin gelişimi anomalisidir. Normalde beyin erken dönemdeki gelişimi sırasında ikiye bölünür (hemisferler). Holoprozensefali, beynin sağ ve sol hemisferlere ayrılacak şekilde bölünmediğinde ortaya çıkar. Bu duruma, genetik sendromların, kromozom anormalliklerinin veya doğumsal kusurlara neden olan maddelerin (teratojenler) neden olduğu diğer holoprozensefali tiplerinden ayırmak için nonsendromik denir. Nonsendromik holoprozensefali’nin şiddeti, aynı aile içindeki bireyler arasında bile değişkendir.Nonsendromik holoprozensefali, beynin bölünme derecesine göre dört tipte gruplanabilir. En şiddetli tipten hafif olana kadar sıralanacak olursa, tipler alobar, yarı-lobar, lobar ve orta interhemisferik varyant (MIHV) olarak bilinir.
Belirti ve Bulgular
Nonsendromik holoprozensefali’nin en şiddetli formlarında, beyin hiç bölünmez. Etkilenen bireylerde, bir merkezi göz (siklopi) (Şekil 1) ve gözün üzerinde yer alan tüp şeklinde bir burun yapısı (proboscis) (Şekil 1) vardır. Şiddetli Nonsendromik holoprozensefalisi olan bebeklerin çoğu, doğumdan önce veya hemen sonra ölür. Daha az şiddetli formlarda, beyin kısmen bölünür ve gözler genellikle birbirine yakındır (hipotelorizm) (Şekil 2). Hasta bireylerin yaşam süresi beklentileri, semptomların şiddetine bağlı olarak değişir.
Şekil 1 (siklopi ve proboscis) Görüntü Kaynağı: https://www.humpath.com/IMG/jpg_holoprosencephaly_0301.jpgŞekil 2 (hipotelorism) Görüntü Kaynağı: https://ghr.nlm.nih.gov/art/large/hypotelorism.jpeg
Nonsendromik holoprozensefalisi olan insanlar genellikle küçük bir kafaya (mikrosefali) sahiptirler (Şekil 3), ancak beyinde artmış kafa büyüklüğüne (makrosefali) neden olan bir sıvı birikimi(hidrosefali) geliştirebilirler. Diğer bulgular arasında ağız tavanında bir açıklık (yarık damak) (Şekil 4) ve bu yarık damağa üst dudağın bir yarık ile ayrılması (yarık dudak) eşlik edebilir. İki yerine bir merkezi ön diş (tek bir merkezi maksiller incisor ) (Şekil 5) ve düz bir burun köprüsü de belirtiler arasında yer alabilir (Şekil 6). Göz küreleri anormal derecede küçük (microphthalmia) veya hiç oluşmamış (anoftalmi) olabilir.
Şekil 3 (mikrosefali) Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/microcephaly.jpegŞekil 4 Görüntü kaynağı: https://www.cdc.gov/ncbddd/birthdefects/cleftlip.htmlŞekil 5 (tek ,büyük bir maksiller kesici diş) Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/single-maxillary-central-incisor.jpegŞekil 6 Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/depressed-nasal-bridge.jpeg
Nonsendromik holoprozensefalili bazı kişilerde başın şakak kısmında bir daralma , yukarı doğru bakan palpebral fissür , büyük kulaklar,kısa burun, ters dönmüş burun delikleri , burun ve ağız arasındaki boşluğun (philtrum) içe çökük ve geniş olması gibi fasiyal semptomlar olabilir. Genel olarak yüzdeki belirtilerin şiddeti, beyin anormalliklerinin şiddetiyle doğrudan ilişkilidir.Ancak, şiddetli fasiyal semptomları olmayan bireylerde de ciddi beyin anormallikleri olabilir. Bazı insanlarda görünür yapısal beyin anormallikleri yoktur ancak bu durumla ilişkili bazı yüz belirtilerine sahiptirler. Bu bireylerin mikroform holoprozensefali olarak bilinen bir bozukluğa sahip oldukları düşünülür ve tipik olarak ciddi şekilde etkilenmiş bir aile üyesinin doğumundan sonra tanı alırlar.
Nonsendromik holoprozensefalisi olan çoğu insanın gelişimsel olarak geriliği ve zihinsel kapasitesinde eksiklikler vardır. Etkilenen bireyler sıklıkla , çeşitli hormonlar üreten ve beynin tabanında yer alan, tam olarak işlevini yerine getiremeyen bir hipofiz bezine sahiptir. Hipofiz disfonksiyonu, hormonların kısmi veya tamamen yokluğuna yol açtığı için, çeşitli bozukluklara neden olabilir. Nonsendromik holoprozensefalisi ve hipofiz disfonksiyonu olan kişilerde en yaygın olarak, sıvı alımı ve idrar atılımı arasındaki dengeyi bozan diyabet insipidus görülür. Beynin diğer bölümlerindeki disfonksiyon; nöbetlere, beslenme güçlüklerine ve vücut ısısında, kalp atış hızında ve solunum düzeninde sorunlara neden olabilir. Eğer kokuyu algılayan beyin kısmı gelişmemiş veya eksikse, koku duyusu azalabilir (hiposmiye) veya tamamen yok olabilir (anosmia).
Görülme Sıklığı
Nonsendromik holoprozensefali, holoprozensefalili tüm vakaların yaklaşık olarak yüzde 25 ila 50’sini oluşturur ve bu da yaklaşık olarak 10,000 yenidoğanda 1’e denk gelmektedir.
Genetik Değişiklikler
Nonsendromik holoprozensefaliye neden olabilecek 14 tane gen bulunmuştur. Bu genler, özellikle beynin ve yüzün şeklini belirleyen ve normal embriyonik gelişim için önemli olan proteinlerin yapılması için gereklidir. Nonsendromik holoprozensefalisi olanların yaklaşık yüzde 25′ inde bu dört genden birinde bir mutasyon vardır: SHH, ZIC2, SIX3 veya TGIF1. Nonsendromik holoprozensefali ile ilişkili diğer genlerdeki mutasyonlar, olguların sadece küçük bir kısmında bulunur. Bu hastalığa sahip birçok bireyde tanımlanmış bir gen mutasyonu yoktur. Bu bireylerdeki bozukluğun nedeni bilinmemektedir.
Beyin, normalde hamileliğin üçüncü ila dördüncü haftasında sağ ve sol hemisferlere ayrılır. İki hemisfere ayıran çizgiyi kurmak için, birçok genin aktivitesi sıkı bir şekilde düzenlenmeli ve koordine edilmelidir. Bu genler; , sağ ve sol hemisferi oluşturacak sinyal proteinlerini yapmak için gerekli talimatları sağlar.
Sinyal proteinleri, gözlerin oluşumu için de çok önemlidir. Erken dönemdeki gelişim sırasında göze dönüşen hücreler, göz alanı olarak adlandırılan tek bir yapı oluştururlar. Bu yapı gelişmekte olan yüzün merkezinde yer almaktadır. SHH geninden üretilen sinyal proteini, göz alanının iki ayrı göze ayrılmasına neden olur. SIX3 geni, göz merceğinin ve retinanın oluşumunda görev alır.
Nonsendromik holoprozensefaliye neden olan genlerdeki mutasyonlar, anormal veya fonksiyonel olmayan sinyal proteinlerinin üretimine yol açar. Doğru sinyaller olmadan, gözler normal olarak oluşamaz ve beyin iki hemisfere ayrılamaz. Gözlerin doğru pozisyonuna geçmemesi durumunda yüzün diğer bölümlerinin gelişimi de etkilenir. Nonsendromik holoprozensefalinin belirti ve bulguları, beynin ve yüzün anormal gelişmesinden kaynaklanır.
Araştırmacılar, tespit edilmemiş olan diğer genetik veya çevresel faktörlerin, nonsendromik holoprozensefalinin şiddetini belirlemede rol oynadığına inanmaktadır.
Kalıtım Paterni
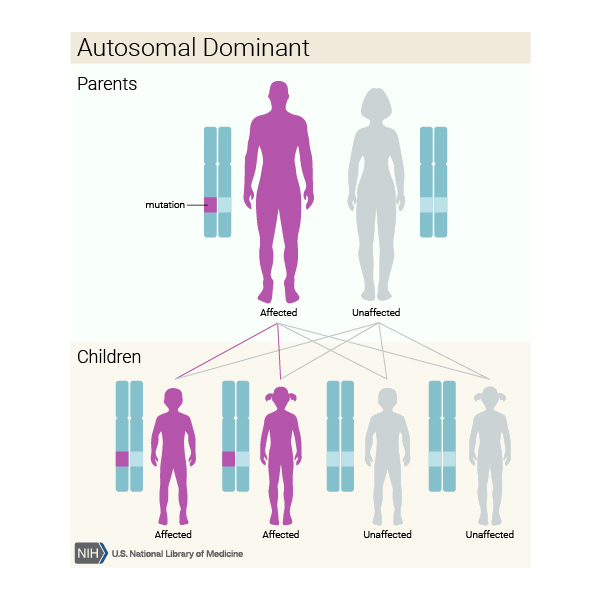
Nonsendromik holoprozensefali otozomal dominant kalıtılır, bu da her bir hücredeki bir genin, bozukluğa neden olmak için genellikle yeterli olduğu anlamına gelir. Bununla birlikte, bir gen mutasyonu olan tüm insanlarda belirti ve semptomlara rastlanılmaz.
Bazı vakalarda, etkilenen kişi, hastalığın özelliklerine sahip olan veya olmayan bir ebeveynin mutasyonunu miras alır. Diğer vakalar ise ailelerinde bozukluk öyküsü olmayan kişilerde, yeni bir gen mutasyonundan kaynaklanır.
Bilinen bir nedeni olmadan HPE’ si olan kişilerin aile üyelerinde hastalığa yakalanma riski genel olarak düşüktür.Sadece bazı durumlarda risk %50 civarı olmaktadır.
Görüntü kaynağı: https://ghr.nlm.nih.gov/art/large/autodominant.jpeg
Tanı Yöntemleri
Çok ciddi vakalar, hamilelik sırasında sistematik ultrason taraması ve manyetik rezonans görüntüleme (MRG) ile tespit edilebilir. Daha sonraki dönemde tanı klinik özelliklere dayanmaktadır.
Ayırıcı Tanı
Ayırıcı tanıda anensefali, ağır konjenital hidrosefali, Walker-Warburg sendromu, geniş interhemisferik kist, otosefali ve diğer orta hat defektleri bulunur.
Antenal Dönemde Tanı
Yüksek klinik değişkenlik nedeniyle prenatal tanı, moleküler tanı yerine ultrason taramaları ve MRG’ ye dayanır.Moleküler tanı diyabetli annelerde ve aile öyküsü olanlarda yararlı olabilir.
Hastalıkla İlişkili Genler
Non sendromik HPE’ de, en az 14 gen etkilenmiştir: 4 majör gen (SHH (7q36), ZIC2 (13q32), SIX3 (2p21), TGIF (18p11)) ve 10 minör gen (PTCH1 (9q22) GLI2 (2q14), FOXH1 (8q24), TDGF1 (3p21), DISP1 (1q42), NODAL (10q22), FGF8 (10q24), GAS1 (9q21), DLL1 (6q27) ve CDON (11q23-q24)).tespit edilmiştir.
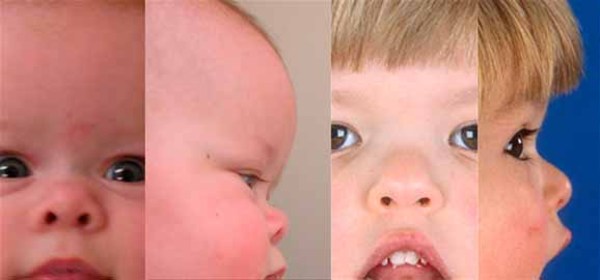
Cornelia de Lange sendromu, vücudun birçok kısmını etkileyen gelişimsel bir bozukluktur. Bu bozukluğun özellikleri etkilenen bireyler arasında oldukça çeşitlilik gösterir ve görece hafif ya da şiddetli olabilir.
Hastalığın karakteristik özellikleri: doğum öncesi ve sonrası yavaş gelişim sonucu kısa boy, orta veya ileri derecede zeka geriliği ve kol, kafa ve parmak kemiklerindeki anormalliklerdir. Ayrıca Cornelia de Lange sendromu olan insanların çoğu; kemerli ve birleşik kaş, uzun kirpikler, aralıklı dişler, yukarı kalkık ve küçük burun ile kulakların başın aşağısında olması gibi belirgin yüz özelliklerine sahiptir. Bu hastalıktan etkilenmiş olan insanların birçoğunda, bir iletişim ve sosyal etkileşim hastalığı olan otizm benzeri davranışsal bozukluklar da görülmektedir.
Fazla miktarda vücut kılı, normalden küçük kafa, işitme kaybı ve sindirim sistemi problemleri de hastalığın belirti ve semptomlarından olabilir. Bu sağlık sorununa sahip insanların bazıları “yarık damak/yarık dudak” denilen ağız tabanında bir açıklıkla doğarlar. Nöbetler, kalp rahatsızlıkları ve göz sorunları da bu hastalığa sahip insanlarda gözlemlenmiştir.
Genetik Değişiklikler
Cornelia de Lange sendromunu, en az beş genin mutasyonu sonucu oluşabilir: NIPBL, SMC1A, HDAC8, RAD21 ve SMC3. NIPBL gen mutasyonu bu hastalığa sahip insanların yarısından fazlasında tespit edilmiş bulunmaktadır, diğer genler ise daha az yaygındır.
Bu beş genden üretilen proteinler, doğum öncesi gelişimde önemli rol oynayan “kohezin kompleksi”nin yapısına katılır veya işlevini yerine getirmesinde yardımcı olur. Kohezin kompleksi, hücre içinde kromozom yapısı ve organizasyonunun düzenlenmesine, hücrenin genetik bilgisini dengelenmesine ve zarar görmüş DNA’nın tamirine yardımcı olur. Bu kompleks aynı zamanda kol, bacak, yüz gibi vücut bölümlerinin gelişimine rehberlik eden genlerin aktivitesini düzenler.
NIPBL, SMC1A, HDAC8, RAD21 ve SMC3 genlerindeki mutasyonlar, kohezin kompleksinin faaliyetini zayıflatarak erken gelişimin önemli dönemlerinde gen regülasyonunu bozar ve Cornelia de Lange sendromuna sebep olurlar.
Cornelia de Lange sendromunun özellikleri oldukça çeşitlilik göstermekte ve aynı gen mutasyonuna sahip bireylerde bile hastalığın şiddeti farklılık gösterebilmektedir. Bilim insanları, fazladan genetik veya çevresel faktörlerin bireye özgü belirti ve semptomların belirlenmesinde önemli olduğunu düşünmektedirler. Genel olarak, SMC1A, RAD21 ve SMC3 gen mutasyonları, NIPBL geni mutasyonuna göre daha hafif şiddette belirti ve semptoma sebep olmaktadır. HDAC8 genindeki mutasyonları ise biraz daha farklı sonuçlar vermektedir. Örneğin: bebeklerde kafadaki yumuşak dokunun geç kapanması (bıngıldak), ayrık gözler ve dental anormallikler. NIBPL gen mutasyonuna sahip bireyler gibi, HDAC8 gen mutasyonuna sahip bireylerde ileri dercede zeka geriliğine sahip olabilirler.
Cornelia de Lange sendromu vakalarının yaklaşık yüzde otuzunun sebebi bilinmemektedir. Bilim insanları bilinen beş gendeki fazladan değişikliklerin ve başka genlerdeki mutasyonların bu hastalığa sebep olabileceğini düşünmektediler.
Kalıtım
Cornelia de Lange sendromu NIPBL, RAD21 veya SMC3 genlerindeki mutasyonlar sonucu oluşsa da, otozomal baskın bir şekilde kalıtıldığı düşünülmektedir. Otozomal baskın kalıtım, hücrelerde bir değişmiş kromozomun olmasının hastalığın ortaya çıkması için yeterli olması demektir. Vakaların çoğu, yeni gen mutasyonlarının sonucu ve hastalık aile geçmişinde bulunmayan kimselerde görülmektedir.
Cornelia de Lange sendromu HDAC8 veya SMC1A genlerinde ortaya çıkan mutasyonla oluştuğunda hastalık X kromozomuna bağlı olarak baskın bir şekilde kalıtılır. Mutasyona uğrayan gen X kromozomu (cinsiyet kromozomlarından biri) üzerinde bulunuyorsa hastalık X-bağımlı olarak kabul edilir. X-bağımlı Cornelia de Lange hastalığı hakkında yapılan araştırmalar, hücrede, değişime uğrayan genin bir adedinin bulunmasının hastalığa sebep olmak için yeterli olduğunu göstermektedir. Erkeklerin kadınlara göre daha sık veya daha şiddetli etkilendiği X-bağımlı çekinik kalıtımın aksine, X-bağımlı baskın kalıtımda erkekler ve kadınlar benzer şekilde etkilenir. Birçok vaka HDAC8 veya SMC1A genlerindeki yeni mutasyonlar sonucu oluşur ve hastanın aile geçmişinde bu hastalık bulunmaz.
Tanı
Moleküler testler ilgili genlerde bir mutasyon tespit etmişse birey CdLS sahibidir. Aksi halde, klinik sonuçları, yüz kriterlerine uymalı, bu kriterler, altı sistem kategorisinden en az iki veya üçüne ait olmalıdır. Sistemlerden en az biri büyüme, gelişme veya davranışla ilgili olmalıdır. Bu kriterlere uyuyorsa bireye klinik olarak CdLS teşhisi konmuştur.
Tanı Koyma
CdLS gen testinde pozitif muyaston VEYA
Yüzsel bulgular ve ana kategorilerden birine giren iki kriter VEYA
Yüzsel bulgular ve ana kategoriye giren bir, fazladan da iki kriter (ana veya yan kategoriye ait)
Yüzsel Bulgular
Birleşik kaş VE >aşağıdakilerden üç veya daha fazlası
Uzun kirpikler
Kısa burun, öne eğik burun delikleri
Üst dudak ve burun arasında uzun ve belirgin alan
Geniş ya da basık burun kemeri
Küçük ya da kare çene
İnce, köşeleri aşağıya dönük dudak
Yüksek damak
Geniş aralıklı ya da eksik diş
Ana Kategori
BÜYÜME >aşağıdakilerden iki veya daha fazlası
Yaşıtlarından yüzde beş daha az ağırlık
Yaşıtlarından yüzde beş daha az boy
Yaşıtlarından yüzde beş daha küçük ölçekte fetüs kafası
GELİŞME >aşağıdakilerden bir veya daha fazlası
Gelişim geriliği veya zeka geriliği, (kas yeteneğindense konuşma becerisi daha çok etkilenmiştir)
Öğrenme zorlukları
DAVRANIŞ >aşağıdakilerden iki veya daha fazlası
Dikkat dağınıklığı ve hiperaktivite
Obsesif-kompülsif davranışlar
Anksiyete
Sürekli dolaşma ihtiyacı
Sinirlilik
Kendine zarar verme
Aşırı utangaçlık ve çekingenlik
Otizm benzeri hareketler
Yan Kategori
KAS VE İSKELET İLE İLGİLİ >aşağıdakilerden bir veya daha fazlası
Eksik kol veya önkol
Küçük el ve/veya ayaklar, el ve ayakta eksik parçalar
Kıvrılmış serçe parmak
Avuç içinde anormal katlanmalar
Çıkık/anormal dirsek
Kısa başparmak eklemi
El kemiğinin başparmakla birleştiği yerde şekil bozukluğu
Ve 3. Ayak parmakları arasında kısmi ağ
Omurga eğriliği
Göğüs veya sternum deformasyonu
Çıkık kalça veya displazi
SİNİRSEL/DERİ >aşağıdakilerden üç veya daha fazlası
Düşük göz kapağı
Gözyaşı kanalı bozukluğu ya da gözkapağı iltihabı
Miyop
Ciddi göz şekil bozukluğu veya retina incelmesi
Sağırlık ya da işitme kaybı
Nöbetler
Alacalı deri
Fazla miktarda vücut tüyü
Küçük göğüs ucu ve/veya göbek deliği
DİĞER ÖNEMLİ SİSTEMLER >aşağıdakilerden üç veya daha fazlası
Sindirim sistemi şekil/konum bozuklukları
Diyafragmatik fıtık
Mide ve yemek borusunda reflü
Yarık damak
Konjenital kalp rahatsızlıkları
Mikropenis
Anormal yerleşmiş üretra açıklığı
Börek/idrar torbası şekil bozuklukları
Tedavi-Kontrol
Cornelia de Lanfe sendromuna sahip olan hastalara yönelik ilgi, yaş ile bağlantılıdır. Bebek ilk, çocukluk, gençlik ve yetişkinlik çağındaki hastaların ihtiyaçları farklı olur.
Bebeklikte ve Tanı Koyulma Zamanında
Potansiyel CdLS tanısı düşünülüyorsa karyotip elde edilmelidir. (Kandaki kromozomlar değerlendirilmelidir). Tanı koyulduktan sonra önerilen hizmetler aşağıdaki gibidir:
Ekokardiyogram
Renal ultrason
Pediyatrik göz muayenesi
İşitme değerlendirmesi (otoakustik emisyon veya beiyin sapının uyarılması ölçülür).
UGI testi (sindirim sisteminin üst kısımlarının – yemek borusu, mide, duodenum- X-Ray ile taranması. Malrotasyon ya da reflü var mı diye bakılır).
Gastraözofogal reflü taraması (ph ölçümü ve/veya endoskopi yapılır. Varsa ilaç veya ameliyatla tedavi edilir)
Gelişme değerlendirmesi (bebeklikte ve sonrasında her 1-3 yılda bir)
Erken müdahale hizmetleri başlar ve ihtiyaçlar karşılana kadar devam eder
Uygun CdLS gelişim tablosu takip edilerek gelişme değerlendirmesi yapılır (genellikle yüksek kalorili besinler önerilir fakat bireyler hızlı metabolizmayla kendi tempolarında büyümeye devam ederler)
Destek veren kurumların iletişim bilgileri ailelere verilir
İleriki hamileliklere yönelik moleküler test önerilir
Erken Çocukluk(Bir İla Sekiz Yaş Arası)
Birey düzenli değerlendirme ve aşılamaya tabi tutulmalıdır.
Erkeklerde yerine inmemiş testisler 18 aya kadar düzetilmelidir
Devam eden gelişim hizmetleri, okul, terapi gibi meseleler bireyselleştirilmelidir. Bireyler fiziksel, mesleki ve konuşma terapilerinden faydalanacaktır. İşaret dili öğrenimi, sözlü iletişimi kolaylaştıracağından, tavsiye edilmektedir.
CdLS gelişim tablolarından bireyin büyüme/gelişmesi takip edilmeye devam edilir.
Altı ayda bir diş muayenesi
İlk değerlendirmenin sonuçlarına göre bir kere veya yıllık pediyatrik göz değerlendirmesi
Her iki-üç yılda bir işitme testi
Sindirim sistemi rahatsızlıkları, reflü ihtimaline karşı sürekli olarak değerlendirilmelidir. Endoskopi en büyük verimi verse de ph ölçeği de düşünülmelidir.
Bağırsak düğümlenmesinin herhangi bir belirtisine karşın direk acile getirilmelidir, ameliyat gerekebilir.
Operasyon gerçekleşirse, sonrasında uzmanlar tarafından takibe alınmalıdır
Herhangi bir ameliyat gerçekleştiğinde, anesteziden daha çok verim alabilmek amacıyla, alakalı uzmanların tamamının görüşleri dikkate alınmalıdır. Böylelikle aynı zamanda birey, ihtiyaç duyduğu şekilde tanısal veya kontrol amaçlı incelenmiş olur.
Bebekliğin j,k maddeleri.
Geç Çocukluk (Sekiz Yaşından Ergenliğe Kadar)
Birey düzenli değerlendirmeye tabi tutulmalıdır.
Eklem kasılmaları, kalça sorunları, nasır, omurga eğriliği ve ortotik şikayetler için ortopedist ile görüşümelidir.
ADHD,kendine zarar verecek şekilde davranışlar gibi sorunlar baş gösterirse davranışsal değerlendirmeye tabi tutulmalıdır.
Erken çocukluğun b,k maddeleri.
Gençlik (Ergenlikten 20 Yaşına Kadar)
Birey düzenli değerlendirmeye tabi tutulmalıdır.
Devam eden gelişim hizmetleri. Okul yerleştirilmesi ve terapi bireyselleştirilmelidir. Okul veya liseden sonra işe yerleştirme, mesleki eğitim ve/veya yükseköğrenim için erkenden plan yapılmalıdır.
Kadınlar için geç ergenlikten başlayarak yetişkinlik süresince, cinsel aktiviteye bağlı olarak, en az üç yılda bir Pap Smear testi ile pelvik muayene düşünülmelidir. Hasta ve ailesiyle adet döngüsü düzeni ve doğum kontrol ile ilgili hormon tedavisi hakkında görüşülmelidir. (hasta ve ailesine göre spesifik)
Gelişimsel olarak yatkınlığı varsa nüks riski hakkında aileye konuşulmalıdır.
Geç çocukluğun a,c maddeleri.
Yetişkinlik
Birey düzenli değerlendirmeye tabi tutulmalıdır.
Kan basıncı takip edilmeli, baseline EKG düşünülmeli, düzenli göğüs, testis ve prostat muayenesi olunmalıdır.
Daha yüksek eğitim, iş meseleleri veya mesleki hazırlık konuşulmalıdır.
ADHD, OCD, kendine zarar verecek davranışlar ve depresyon gibi durumlar ortaya çıkarsa davranışsal veya psikiyatrik değerlendirme yapılmalıdır
Osteoporoz oluşup oluşmadığını anlamak için DEXA scan kullanılmalıdır
Şikayetlere göre her 4-6 ayda bir, özel ihtiyaçları olan ailelere aşina dişçiler tarafından diş muayenesi olunmalıdır
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Fabry hastalığı lizozomal depo hastalığıdır. Lizozomlar yuvarlak, içi enzim denen özel proteinlerle dolu yapılardır. Lizozomal enzimler diğer proteinlerin, karbonhidratların yağların ve diğer yapıların yıkımına sebep olur. Bu hastalıkta alfa-galaktosidaz (α-GLA) enziminin yeterli miktarda olmamasından kaynaklanır.GLA genindeki mutasyon bu hastalığa sebep olur.GLA geni α-GLA enzimini kodlar. α-GLA enzimi globotriaosilseramidin(GL3) yıkımına sebep olan bir enzimdir. Bu enzimin eksikliği, lizozozomda GL3 birikmesine sebep olur. Bu birikme de hücre anomalilerine, organlarda (özellikle küçük kan damarlarında, kalpte ve böbreklerde) işlev bozukluğuna sebep olur. Semptomlar hastadan hastaya farklılık gösterebilir. Hastalığın daha hafif seyreden versiyonu da vardır.
Belirti ve Semptomlar
Hastalığın semptomları arasında özellikle ellerde ve ayaklarda hissedilen yanma hissi, anjiyokeratoma olarak adlandırılan ciltte görülen küçük siyah kırmızı noktalar, terlemede azalma, korneada bulanık bir görüntü oluşmasına sebep olan kornea distrofisi, midede ağrı, kireçlenme, düşük kırmızı kan hücresi miktarı görülebilir. Aynı zamanda kalp böbrek ve beyni de etkileyebilir, inme, kalp krizi ve böbrek yetmezliği de bu sebepten görülebilir. Ek olarak; hipertansiyon, ishal, baş ağrısı, kronik yorgunluk, güç kaybı, mide bulantısı/kusma, geciken ergenlik, nadir olarak eklem yerlerinde malformasyon görülmesi gibi belirti ve semptomları vardır.
Genetik Görülme Sıklığı
Bu hastalığın görülme sıklığı erkeklerde 1/40.000 olarak hesaplanmıştır. Aynı zamanda kadınlarda da görülmektedir fakat görülme sıklığı henüz bilinmemektedir.
Kalıtım Paterni/Deseni
Fabry hastalığı X’e bağlı kalıtımla aktarılır. X’e bağlı kalıtımda, hastalığa sebep olan gen X kromozomunda bulunur. Erkeklerde bir tane X kromozomu bulunduğu için GLA genindeki mutasyon bu hastalığın oluşmasına sebep olur. Kadınlarda iki adet X kromozomu bulunduğu için bir kopyadaki GLA geninde oluşan mutasyon genelde hastalığın daha hafif semptom gösteren formunun görülmesine hatta hiç semptom görülmemesine sebep olabilir. Sadece bir kopyada mutasyona sahip olan kadın bireyler Fabry Hastalığı için taşıyıcı veya heterozigot olarak adlandırılır. Eğer anne Fabry hastalığı için taşıyıcı ise, baba da sağlıklı ise çocuğun bu hastalığa sahip olma riski cinsiyetine bağlıdır. Çocuk erkekse %50 şansla bu hastalığa sahip olabilir, %50 şansla etkilenmeyebilir. Eğer çocuk kızsa %50 şansla taşıyıcı olabilir veya %50 şansla etkilenmeyebilir. Eğer babada mutasyon görülüyorsa fakat anne sağlıklı ise bütün erkek çocukları sağlıklı, bütün kız çocukları da taşıyıcı olur.
Teşhis Yöntemleri ve Tedaviler
Hastalık enzim analizi (alfa galaktosidaz enzimi için), genetik test, karakteristik bulgularla teşhis edilebilir.
Fenitoin, karbamazepin, gabapentin; el ve ayaklarda hissedilen yanma hissi için kullanılabilir. ACE inhibitörleri böbrek yetmezliği durumunda kullanılabilir. Eğer bu yeterli gelmezse böbrek nakli yöntemine başvurulabilir. Bu durumda hastanın böbreğindeki GL3 birikimi duracaktır fakat Fabry hastalığının diğer organlar üzerindeki etkisi devam edecektir. Böbrek nakli yapılmadan önce donörün taşıyıcı veya henüz hastalığı teşhis edilmemiş bir birey olmamasına dikkat edilmelidir. Enzim replasman tedavisi aynı zamanda bazı semptomların etkisini azaltmak için kullanılabilir. Fakat bu yöntem uzun vadede sonuç elde edilmek istendiği zaman kullanılabilir. Kan basıncını ve kolesterol seviyesini düzenlemek için çeşitli ilaçlar kullanılablir. Aspirin kalp krizini önlemek için kullanılabilir.Galafold da aynı zamanda missens mutasyonların etkisini enzimatik aktiviteyi arttırarak azaltabilir. Fabry hastalığına sahip olan bireyler ve aileleri için genetik danışmanlık da önerilmektedir.
(Bu yazıda diskeratosis konjenitanın özellikleri anlatılmıştır. Revesz sendromu bu hastalığın bir varyasyonudur.)
Tanım
Diskeratosis konjenita vücudumuzun birçok bölgesine etki eden bir hastalıktır.Bu hastalığın 3 karakteristik özelliği vardır: ayak ve el tırnaklarının gelişimindeki zayıflık veya şekillerindeki anormallik/tırnak distrofisi ; boyun ve göğüste cilt renginde değişiklikler (pigmentasyon), genellikle “dantelli” olarak tanımlanan bir modelde; Ve ağızda beyaz lekeler (oral lökoplaki).
Diskeratosis konjenita olan insanlar yaşamı tehdit eden birçok durum geliştirme riski taşırlar. Özellikle kemik iliği fonksiyonlarını bozan rahatsızlıklara karşı savunmasızdırlar. Bu bozukluklar, kemik iliğinin yeni kan hücreleri üretme kabiliyetlerini aksatır. Etkilenen kişiler kemik iliği yetmezliği olarak da bilinen aplastik anemi oluşturabilirler; bu da kemik iliği yeterince yeni kan hücresi üretmediğinde ortaya çıkar. Ayrıca, olgunlaşmamış kan hücrelerinin normal olarak gelişemediği bir durum olan miyelodisplastik sendrom için ortalama riskin de üstündedirler; Bu durumda lösemi adı verilen bir kan kanseri gelişim gösterebilir. Diskeratosis konjenita’lı insanlarda miyelodisplastik sendrom gelişmese dahi lösemi geliştirme riski yüksektir. Buna ek olarak, diğer kanserler, özellikle baş, boyun, anüs veya cinsel organ kanserlerinin gelişme riskinden daha yüksektir.
Diskeratosis konjenita’lı kişiler akciğer fibrozisi geliştirebilirler; bu durum akciğerlerde, gelişmekte olan bir yara dokusunun oluşumuna neden olur ve kan dolaşımında oksijen taşınmasını azaltır. Diskeratosis konjenita’lı bazı insanlarda ortaya çıkan ek bulgular ve belirtiler arasında göz yorgunluğuna yol açan tıkanmış dar gözyaşı kanalları, gözyaşı akmasını önleme ve göz kapaklarının tahriş olmasına yol açan göz anormallikleri; Diş problemleri; Saç dökülmesi veya erken gri saç; Düşük kemik mineral yoğunluğu (osteoporoz); Kalça ve omuz eklemlerinde dejenerasyon (avasküler nekroz); Veya karaciğer hastalığı. Bazı etkilenen erkeklerde idrarını mesaneden vücudun dışına çıkaran tüp olan üretranın daralması (stenoz) olabilir. Üretral darlık, zor ya da ağrılı idrar ve idrar yolu enfeksiyonlarına neden olabilir. Diskeratosis konjenita’nın şiddeti, etkilenen kişiler arasında büyük farklılıklar göstermektedir. En az etkilenen bireyler, bozukluğun hafif fiziksel özelliklerine ve normal kemik iliği işlevine sahiptir. Daha şiddetli şekilde etkilenen bireyler karakteristik fiziksel özelliklerin çoğuna sahiptir ve erken yetişkinlikte kemik iliği yetmezliği, kanser veya pulmoner fibroz (akciğer fibrozisi) deneyimindedir. Diskeratozis konjenitanın görüldüğü birçok insan normal zeka ve gelişmiş motor becerilerine (ayakta durmak ve yürümek gibi) sahipken, bazı ciddi etkilenen bireylerde gelişimsel gecikme görülebilir. Diskeratosis Konjenita’nın şiddetli türlerinden biri olan Hoyeraal Hreidaarsson sendromunda , etkilenen bireyler alışılmışın dışında küçük ve gelişmemiş serebelluma( hareketi koordine eden beynin bir parçası) sahiptir. Revesz sendromu olarak adlandırılan bir diğer ciddi varyant ise, diskeratosis konjenitanin diğer semptomlarına ek olarak gözün arka kısmındaki (retinadaki) ışığa duyarlı dokudaki anormallikleri de içerir.
Sıklık
Diskeratosis konjenita’nın tam prevalansı bilinmemektedir. Yaklaşık olarak milyonda 1 gerçekleştiği tahmin edilmektedir.
Genetik Değişiklikler
Diskeratosis konjenita olan kişilerin yaklaşık yarısında hastalık, TERT, TERC, DKC1 veya TINF2 genindeki mutasyonlardan kaynaklanır. Bu genler, kromozomların uçlarında bulunan telomerler olarak bilinen yapıların korunmasına yardımcı olan proteinlerin yapılması için talimatlar sağlar. Diskeratosis konjenita ya sahip olan az sayıdaki bireyde, telomer korunumuyla ilgili diğer genlerdeki mutasyonlar tespit edilmiştir. Diğer etkilenen bireylerin şu anda diskeratosis konjenita ile ilişkili genlerin herhangi birinde mutasyonu yoktur. Bu vakalarda, bozukluğun nedeni bilinmemekle birlikte, telomer korunumu ile ilgili diğer tanımlanamayan genler muhtemelen söz konusudur. Telomerler, kromozomların anormal olarak birbirine yapışmasını veya parçalanmasını engeller.Birçok hücrede, telomer, hücre bölünürken kademeli olarak kısalır. Belli sayıda hücre bölünmesinden sonra, telomerler o kadar kısa olur ki hücreyi bölünmesini durdurmak veya kendini yok etmesi için tetikler (apoptozise uğrar). Telomerler, telomeraz ve şelterin adı verilen iki önemli protein kompleksi tarafından idame ettirilir. Telomeraz, her hücre bölündüğünde kromozomların uçlarına DNA’nın küçük tekrarlanan bölümlerini ekleyerek normal telomer uzunluğunu korumaya yardımcı olur. Telomerazın ana bileşenleri olan hTR ve hTERT, sırasıyla TERC ve TERT genlerinden üretilmektedir. HTR bileşeni bir RNA molekülüdür, DNA’nın kimyasal bir kuzenidir. Telomerazın kromozomların uçlarına eklediği tekrarlanan DNA dizisini oluşturmak için bir şablon sağlar. HTERT bileşeninin işlevi, yeni DNA segmentini kromozomun uçlarına eklemektir. DKC1 geni, telomeraz fonksiyonunda önemli olan bir başka protein yapmak için talimatlar sağlar. Dispersin adı verilen bu protein, hTR’ye bağlanır (bağlar) ve telomeraz kompleksinin dengede kalmasına yardımcı olur. şelterin kompleksi hücrenin DNA onarım sürecinden telomerleri korumaya yardımcı olur. Korunma mekanizması olmadan, onarım mekanizması, kromozomal uçları DNA dizisinde anormal kopmalar olarak algılar ve uçlara birlikte katılmaya veya apoptozu başlatmaya çalışır. TINF2 geni, shelterin kompleksinin bir parçası olan bir protein yapmak için talimatlar sağlar. TERT, TERC, DKC1 veya TINF2 gen mutasyonları, telomerazın bozulmasına ve telomer uzunluğunun azalmasına neden olan telomeraz veya shelterin komplekslerinin işlev bozukluğuna neden olur. Hızla bölünen hücreler özellikle kısaltılmış telomerlerin etkilerine karşı savunmasızdır. Sonuç olarak, diskeratosis konjenita’lı insanlar tırnak yatakları, kıl follikülleri, deri, ağız astar (oral mukoza) ve kemik iliği gibi hızlı bölünen hücrelerinde çeşitli problemlerle karşılaşabilirler.
Telomer korunumunun yetersizliğinden kaynaklanan kromozomların kırılması ve kararsızlığı, hücrelerin kontrolsüz bir şekilde bölünmesine izin veren genetik değişikliğe neden olabilir ve bu da diskeratosis konjenita’lı kişilerde kanser gelişimine neden olur.
Kalıtım Şekilleri
Diskeratosis konjenita farklı kalıtım kalıplarına sahip olabilir.
Diskeratosis konjenita’ya DKC1 gen mutasyonları neden olduğunda, hastalık X-bağlı resesif kalıptan miras kalır. DKC1 geni, iki cinsiyet kromozomundan biri olan X kromozomu üzerinde bulunur. Erkeklerde (Sadece bir X kromozomuna sahipler), her hücrede genin değiştirilmiş bir kopyası hastalığa sebebiyet vermesi için yeterlidir. Dişilerde (iki X kromozomu), bozukluğa neden olmak için genin her iki kopyasında da bir mutasyon meydana gelmelidir. Dişilerin bu genin değiştirilmiş iki kopyasına sahip olması muhtemel olmadığı için, erkekler X’e bağlı resesif bozukluklardan kadınlardan daha sık etkilenirler. X’e bağlı kalıtımın karakteristik bir özelliği, X e bağlı genlerin babadan oğula geçememesidir.
Diskeratosis konjenita, diğer genlerdeki mutasyonlardan kaynaklandığında, otozomal dominant veya otozomal resesif kalıptan miras alınabilir. Otozomal dominant, her bir hücredeki değiştirilmiş genin bir kopyasının bozukluğa neden olması için yeterli olduğu anlamına gelir. Otozomal resesif, her bir hücredeki her iki kopyanın da mutasyona uğradığı anlamına gelir. Otozomal resesif durumda olan bir bireyin ebeveynleri mutasyona uğramış genin bir kopyasını taşır, ancak genellikle durumun bulgu ve belirtilerini göstermezler.