Burn-McKeown sendromu (BMKS) kromozom 18q23 üzerindeki TXNL4A geninde (611595) homozigot veya bileşik heterozigot mutasyondan kaynaklandığına dair kanıtlar ile genetik geçişli bir hastalıktır.
Genetik Değişiklikler / Etken Faktörler
Burn-McKeown sendromu, TXNL4A genindeki mutasyonlardan veya genin protein üretimini kontrol eden promoter bölgesi olarak adlandırılan TXNL4A geninin yakınındaki bir alandan kaynaklanır. TXNL4A geni, insan hücrelerinde bulunan iki spliceosom tipinden daha büyük olan majör spliceosome adı verilen bir protein kompleksinin bir parçasının (alt birimi) yapılması için talimatlar sağlar. Spliceosomes, proteinleri üretmek için genetik bir plan görevi gören DNA’nın kimyasal kuzeni olan mesajcı RNA (mRNA) işlemesine yardımcı olur. Spliceosomes, olgun mRNA’nın üretilmesine yardımcı olmak için olgunlaşmamış mRNA moleküllerinden gelen intronlar olarak adlandırılan bölgeleri tanır ve çıkarır.
Burn-McKeown sendromuna neden olan TXNL4A genini etkileyen mutasyonlar, genden üretilen protein miktarını azaltır. Araştırma, bu spliceosome alt ünitesinin azalan miktarlarının majör spliceosome takımını etkilediğini ve belirli bir mRNA molekülü grubunun üretimini değiştirdiğini düşündürmektedir. Bu değişikliklerin detayları ve Burn-McKeown sendromunun spesifik belirtileri ve semptomları ile ilişkisi bilinmemektedir. Bununla birlikte, spliceosome formasyonu veya fonksiyonuna dahil olan çeşitli genlerdeki mutasyonların, kafa ve yüze (kraniyofasiyal malformasyonlar) etki eden anormalliklere sahip başka koşullara neden olduğu gösterilmiştir, bu nedenle kranyofasiyal gelişimin spliceosome problemlerine özellikle duyarlı olduğu düşünülmektedir.
Semptomlar
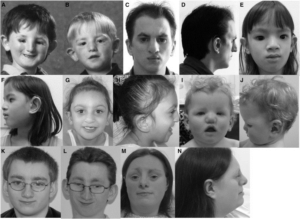
Burn-McKeown sendromu, normal entellektüel gelişimi olan bireylerin, koanal atrezi (Burnun doğumsal tıkanıklığı), sensorinöral sağırlık (Sinirsel işitme kaybı), kardiyak defekt (Kalbin pompalama işlemindeki eksiklik-kusur) ve dar palpebral fissürlerden (Derin yerleşimli gözler, dar gözkapağı kıvrımı) oluşan tipik kraniyofasiyal dismorfizm (Baş ve yüz anomalileri), alt göz kapaklarının kolobomu (kusurlu gelişimi veya yokluğu), yüksek burun ile belirgin burun, yarık dudak ve/veya damak, büyük ve çıkıntılı kulaklar gibi karakteristik kombinasyonlarla çok nadir görülen bir konjenital (doğumsal) anomali durumudur. Akıl genellikle normaldir.
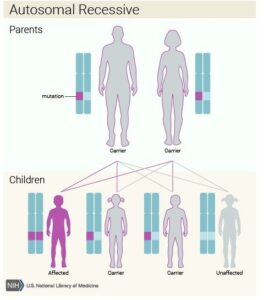
Kalıtım Deseni
Çekinik geçişli bir rahatsızlığı olan bireyin, anne babalarının her biri olumsuz yönde değişmiş (mutasyona uğramış) genin bir kopyasını taşır, ancak tipik olarak durumun belirti ya da semptomlarını göstermezler.
Teşhis Yöntemleri Ve Tedaviler
Belirtilerin tedavisi: Doğumda hava yolu uzaması olan yenidoğanlarda koanal stenoz / atrezinin entübasyon veya cerrahi olarak düzeltilmesi gerekebilir. Korneal maruziyete neden olabilecek alt göz kapaklarının kusurları, bir göz doktorunun korneal skarlaşma riskini azaltmak için müdahalesini gerektirebilir. İşitme kaybının tedavisinde geleneksel işitme cihazları kullanılabilir. Kraniyofasiyal manifestasyonların tedavisi (örneğin yarık dudak ve / veya damak, preauriküler etiketler, belirgin kulaklar), multidisipliner bir ekip tarafından yönetilir. Kardiyak defektler rutin olarak kontrol edilir. Baş ve yüz anomalileri, bozukluklarda uzmanlığa sahip bir hekim tarafından gelişim izlenerek; boy ve kilo rutin ölçümleri yanında, işitme değerlendirmesi ve oftalmolojik (Görme yolları hastalıkları ve cerrahisi) muayene ve cerrahi işlemlerle takip edilmelidir.
Hastalığın Diğer İsimleri
- BMKS, OOFD
- Kraniofasiyal disformizm sendromu
- İki taraflı koanal atrezi, sağırlık ve disformik görünüm
- Okülo-oto-yüz displazisi
Kaynaklar
https://ghr.nlm.nih.gov/condition/burn-mckeown-syndrome
https://www.rightdiagnosis.com/b/burn_mckeown_syndrome/intro.htm