Diğer İsimleri
- Aspartilglukosamınurya
- Aspartilglukosamidaz (AGA) eksikliği
- AGU
- AGA eksikliği
- Glikosilasparaginaz eksikliği
Genel Tanım
Aspartilglukosaminüri , merkezi sinir sistemini kapsayan ve iskelet anormalliklerine ve bağ dokusu lezyonlarına neden olan ciddi otozomal resesif lizozomal depo bozukluğudur. En karakteristik özelliği ilerleyici zihinsel geriliktir. Bozukluğa, lizozomal enzim glikosilasparaginazın yetersiz aktivitesi neden olur; AGU, genellikle Finlandiya hastalık mirası olarak adlandırılan bozukluklar grubuna aittir.
Belirtiler
Aspartilglikozaminüri olan bebekler doğumda sağlıklı görünür ve gelişim çocukluk boyunca tipik olarak normaldir. 2 veya 3 yaşlarında ortaya çıkan bu durumun ilk işareti genellikle konuşma gecikmesidir. Hafif zihinsel engelli belirtisi daha sonra belirginleşir ve öğrenme yavaş bir hızda gerçekleşir. Entelektüel sakatlık ergenlik döneminde giderek kötüleşir. Bu bozukluğu olan çoğu insan öğrendikleri konuşma yeteneğinin çoğunu kaybeder ve etkilenen yetişkinlerin kelime dağarcığında genellikle sadece birkaç kelime vardır. Aspartilglukozaminüri olan yetişkinlerde nöbet veya hareketle ilgili sorunlar gelişebilir. Bu durumdaki insanlarda giderek zayıflayan ve kırılmaya müsait hale gelen kemikler (olabilirosteoporoz), alışılmadık derecede geniş bir eklem hareketi ( hipermobilite ) ve gevşek cilt semptomları görülür. Etkilenen bireyler, geniş aralıklı gözler ( oküler hipertelorizm ), küçük kulaklar ve dolgun dudaklar içeren karakteristik bir yüz görünümüne sahiptir . Burun kısa ve geniştir ve yüz genellikle kare şeklindedir. Bu duruma sahip çocuklar yaşları için uzun olabilir, ancak ergenlik döneminde büyüme hamlesi olmaması tipik olarak yetişkinlerin kısa olmasına neden olur. Etkilenen çocuklar da sık sık üst solunum yolu enfeksiyonu geçirme eğilimindedir. Aspartilglukozaminüri olan bireyler genellikle yetişkinliğin ortalarına kadar hayatta kalırlar. İskelet deforme olabilir. Omurga bükülmüş olabilir (skolyoz) ve boyun alışılmadık derecede kısa olabilir. Gözler de katarakt gelişebilir. Davranış sorunları yaygındır. Akciğer, kalp ve kan problemleri sonraki yıllarda ortaya çıkma eğilimindedir. Bireylerde %80-%90 görülen belirtiler;
- amino asit metabolizmasında anormallik
- idrarda yüksek aspartilglukozamin seviyeleri
- Gecikmiş konuşma ve dil gelişimi
- İstemsiz kas hareketleri bozukluğu
- Dişeti büyümesi
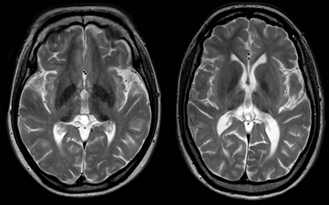
[1.1] İki aspartilglikozaminüri hastası 3.0 T olarak görüntülendi. Solda 34 yaşında kadın hasta, sağda 26 yaşında kadın hasta. Pulvinar çekirdeklerde tipik sinyal yoğunluğu azalması gösteren eksenel T2 ağırlıklı görüntüler. Her ikisinde de, gri ve beyaz madde arasındaki zayıf farklılaşma, sinyal yoğunluğu artışı olan bazı düzensiz joktakortikal odaklar, 3. ventrikülün hafif dilatasyonu, bir epifiz kisti ve nispeten kalın bir kafatası da görülebilir.
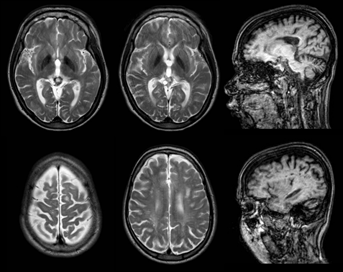
[1.2] 33 yaşında kadın aspartilglikozaminüri hastası, 3.0 T’de görüntülendi. Solda, daha önce aspartilglikozaminüri ‘da bildirilmeyen talamide atipik sinyal yoğunluğu artışı gösteren dört eksenel T2 ağırlıklı görüntü ve periventriküler ve juktakortikal beyaz maddede sinyal yoğunluğu artışı olan birkaç düzensiz odak var. Sağda pulvinar çekirdeğindeki SI azalmasını ve beyaz maddedeki bazı fokal hipertansiyon lezyonlarını gösteren iki sagital T1 ağırlıklı görüntü vardır. Ayrıca bu hastanın görüntülerinde hafif lateral ve 3. ventrikül dilatasyonu, gri ve beyaz madde arasındaki zayıf farklılaşma, hafif dilate perivasküler boşluklar, pineal kist ve hafif genel atrofi görülebilir.
Nedenler
AGA genindeki mutasyonlar aspartilglukosaminüriye neden olur. AGA geni, aspartylglucosaminidase olarak adlandırılan bir enzim üretmek için talimatlar içerir. Bu enzim, geri dönüşüm merkezleri olarak işlev gören hücrelerdeki lizozomlarda aktiftir. Lizozomlar içindeki enzim, belirli proteinlere (glikoproteinler) bağlı şeker molekülleri (oligosakkaritler) komplekslerinin parçalanmasına yardımcı olur. AGA gen mutasyonları, lizozomlarda aspartilglukosaminidaz enziminin yokluğuna veya eksikliğine neden olarak glikoproteinlerin normal parçalanmasını önler. Sonuç olarak, lizozomlar içinde glikoproteinler birikebilir. Fazla glikoproteinler hücrenin normal fonksiyonlarını bozar ve hücrenin tahrip olmasına neden olabilir. Bir glikoprotein birikimi özellikle beyindeki sinir hücrelerini etkilemektedir; bu hücrelerin kaybı aspartilglukosaminüri belirtilerinin ve semptomlarının çoğuna neden olur .
Epidemiyolojisi
Aspartilglikozaminüri ‘nın Finlandiya’daki 18.500 kişiden 1’ini etkilediği tahmin edilmektedir. Bu durum Finlandiya dışında daha az yaygındır, ancak oranı bilinmemektedir.
Kalıtım Kalıbı
Bu durum otozomal resesif paternde kalıtsaldır yani her hücredeki genin her iki kopyasının mutasyonları vardır. Otozomal resesif koşulu olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını göstermezler.
Teşhis Yöntemleri
Biyokimyasal olarak, bir amino asit veya oligosakkarit kromatografisi üzerine aspartilglikozaminin çokça bulunduğu idrarın atılımı ile karakterizedir. Sonuçlar, lenfositler, fibroblastlar, amniyositler veya trofoblastta ölçülen düşük aspartilglikozaminidaz aktivitesi ile doğrulanır.
Tedavi
Bu güne kadar tedavi edici tek girişim, 5 Fin hastası ile sınırlı allojenik kemik iliği aşılama işlemiydi.
Kaynaklar
https://www.researchgate.net/figure/Two-aspartylglucosaminuria-patients-imaged-at-30-T-On-the-left-is-a-34-year-old-female_fig2_277326017 [1.1] fotoğrafın kaynağı
https://www.researchgate.net/figure/A-33-year-old-female-aspartylglucosaminuria-patient-imaged-at-30-T-On-the-left-are_fig1_277326017 [1.2] fotoğrafın kaynağı