Benign (iyi huylu) intrahepatik kolestaz hastalığı, çoğunlukla kronik karaciğer hasarına kadar ilerleme göstermeden belirli aralıklarda gözlemlenen intrahepatik kolestaz atakları ile karakterize olmuş bir kalıtsal karaciğer hastalığıdır. İntrahepatik kolestaz; safra sekresyonunun herhangi bir aşamasında ortaya çıkan bir bozukluk neticesinde toksik maddelerin hücre içine birikmesi ile kolestatik karaciğer hasarına yol açmasıyla oluşmaktadır. BRIC genetik bir hastalık olarak BRIC1 ve BRIC2 olarak iki farklı türe ayrılmaktadır yalnız hastaların semptomları her iki türde de aynıdır. Hastalık benign (iyi huylu) olduğundan karaciğere herhangi bir hasar vermemektedir. Karaciğer disfonksiyonu atakları kalıcı karaciğer hasarına sebep olan progresif ailevi intrahepatik kolestaz (PFIC)’a dönüşebilmektedir.
Belirti ve Semptomlar
Bu hastalıkta gözlemlenen semptomlar genellikle genç yaşlarda veya yirmili yaşlarda olmaktadır. Başlangıçta gelen bir atak ile şiddetli kaşıntı denilmekte olan prutius ve ardından birkaç hafta sonra gözlerde sararma meydana gelmektedir. Bu belirtilere ek olarak halsizlik, sinirlilik, bulantı, kusma ve iştahsızlık belirtileri de görülmektedir. BRIC vücutta yağ emiliminin azalmasına neden olduğundan kaynaklı gaita da yağ atılımı fazla olur ve bunun sonucunda da kilo verimi gerçekleşir.
Çoğu hastalık için semptomlar insandan insana farklılık göstermektedir ve aynı hastalığa sahip insanlar aynı belirtileri göstermeyebilir. Hastalıkla ilişkili semptomların belirlendiği İnsan Fenotip Ontoloji (Human Phenotype Ontology (HPO)) isimli veritabanın da düzenli olarak semptomlar güncellenmektedir. İyi Huylu İntrahepatik Kolestaz hastalığına ilişkin tablo aşağıdaki gibi yer almaktadır.
Tablo 1 : Hastalığa ilişkin semptomlar ve HPO Kimliği
| Semptomlar | HPO Kimliği |
| Akolik gaita/Kil renkli gaita | 0011985 |
| İştahsızlık | 0002039 |
| Kolestatik karaciğer hastalığı | 0002611 |
| Yükseltilmiş hepatik transaminaz/Yüksek karaciğer enzimleri | 0002910 |
| Yorgunluk | 0012378 |
İnsanların %80-%99’unun bu belirtileri bulunmaktadır.
Genetik Görülme Sıklığı
BRIC (İyi Huylu İntrahepatik Kolestaz), nadir görülen bir hastalıktır. Dünya çapında yaklaşık olarak 50.000 ila 100.000 kişiye etki etmekte olan PFIC hastalığından daha az yaygın olduğu bilinmektedir.
Hastalığı Etkileyen Nedenler
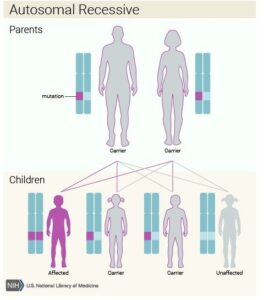
BRIC genetik bir hastalık olarak BRIC1 ve BRIC2 olarak iki farklı türe ayrılmaktadır ve safra salgılanmasına yardımcı olan genler tarafından kontrol edilmektedir. ATP8B1 genindeki mutasyonlar BRIC1 türüne, ABCB11 genindeki mutasyonlar da BRIC2 türüne yol açmaktadır. Otozomal resesif bir şekilde kalıtılmaktadır ve bu bireylerin ebeveynleri taşıyıcıdırlar.
ATP8B1 geni; belli proteinlerin yapımını sağlayarak yağların dağılımını kontrol etmeye yardımcıdır. Bu gendeki gelişen mutasyonlar sonucu safra asitlerinin birikimine yol açmaktadır ve bu da BRIC hastalığına neden olmaktadır.
ABCB11 geni; safra tuzu verme pompası diye adlandırılan BSEP, proteininin yapımını sağlamaktadır. Karaciğerde bulunan ve safra tuzlarının uzaklaştırılmasını sağlayan bir proteindir. Bu gendeki mutasyonlar BSEP fonksiyonunun azalmasına neden olarak BRIC hastalığına neden olmaktadır.
Bazı BRIC hastalığına sahip insanlarda bulunan bu genlerde mutasyon olmamaktadır ve bunun nedeni bilinmemektedir.
Hastalığa İlişkin Teşhis ve Tedavi Yöntemleri
Genetik ve nadir hastalıklarda tanı belirlemek oldukça zordur. Hekimler, kişinin tıbbi geçmişine, semptomlarına, fiziksel muayenesine ve bununla birlikte laboratuvar testlerine bakarak tanıyı belirleyebilirler. Gerektiğinde karaciğer biyopsisi de yapılabilmektedir.
Hastalığın tedavisine ilişkin, rifampisin ve kolestiramin, pruritusu azaltma işlemi gerçekleştirilir. Plazmaferez /MARS (Moleküler Adsorbanlar Sirkülasyon Sistemi)’ında bazı durumlarda fayda sağladığı gözlemlenmiştir. Tıbbı tedavi sonucu yanıt alınmayan hastalar için endoskopik nazobilier drenaj etkili bir yöntemdir. Nazobilier drenaj da hastanın safra kanalına bir kateter yerleştirilerek tedavi süreci boyunca çıkartılmadan kalınır. Genellikle safra taşı ve safra yaralanmalarında kullanılmaktadır ve bu süreçte de safra taşı çıkarılana ve yara iyileşene dek kateter bekletilmektedir. Tabi buna ek olarak kısmi dış biliyer diversiyon da yaşam standartlarını en yüksek seviyeye çıkarmakta ve progresyonu önlemek içinde kullanılmaktadır. Çok fazla atak geçirmekte ve ciddi travmalarda da karaciğer transplantasyonu gerçekleştirmektedir.
Hastalığın Diğer İsimleri
- ABCB11 ilişkili intrahepatik kolestaz
- ATP8B1 ilişkili intrahepatik kolestaz
- BRIC
- Düşük gama-GT ailesel intrahepatik kolestaz
- Tekrarlayan ailevi intrahepatik kolestaz
Kaynakça
- İdilman R. (2006). İntrahepatik Kolestaz Nedenleri ve Tanı Yöntemleri. Türkiye Klinikleri Dahili Tıp Bilimleri Dergisi. 2(34):22-7.
- Knisely A.S., Bull L.N. ve Shneider B.L. (2014). ATP8B1 Eksikliği. GeneReviews(İnternet).
- https://www.ncbi.nlm.nih.gov/medgen/435857
- https://rarediseases.info.nih.gov/diseases/10028/benign-recurrent-intrahepatic-cholestasis-1
- https://ghr.nlm.nih.gov/condition/benign-recurrent-intrahepatic-cholestasis#resources
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=10872&Disease_Disease_Search_diseaseGroup=Benign-recurrent-intrahepatic-cholestasis&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Benign-recurrent-intrahepatic-cholestasis&title=Benign%20recurrent%20intrahepatic%20cholestasis&search=Disease_Search_Simple
- https://rarediseases.info.nih.gov/diseases/12185/benign-recurrent-intrahepatic-cholestasis