Dudak damak yarıklığı hastalığına sahip nadir bir bireysiniz. Dudak damak yarıklığı hastalığı genetik kaynaklı mı?
Tamamen anne karnında gelişimi tamamlayamamaktan kaynaklanan bir durum. Bu stres kaynaklı da olabilir, genetik kaynaklı da olabilir.
Peki dudak damak yarıklığı ilerleyici bir hastalık mı?
Değil. Olabilecek en yüksek noktada doğuyorsun. Gelişimi tamamlayamadığın için eksik doğuyorsun, daha sonrasında hayatında bunun etkilerini görüyorsun.
Tanı koyulurken zorluk yaşandı mı diye sormak istiyorum aslında ama direkt olarak doğum anında fark edilen bir hastalık olsa gerek. Bu süreci anlatır mısınız?
Benim hastalığım doğum anında fark edilmemiş. Normalde doğum anında kontrol edilmesi gereken bir durum fakat beni kontrol etmemişler. Ben bir ila iki hafta kadar anne sütü içememişim çünkü bebeklerde olan ağız refleksini gerçekleştiremiyormuşum. Bu durum ölüme kadar gidebilirdi. Sonrasında annemin, ağzımın içinin simsiyah olduğunu fark etmesiyle birlikte beni doktora götürmüşler ve ancak o zaman anlaşılmış. Ama dediğim gibi, normalde doğumdan hemen sonra konulması gereken bir tanı.
Herhangi bir tedavisi var mı? Eğer varsa tedavileri sınırlı mı?
Tedavisi var. Ameliyat olmak zorundasın çünkü hayatına damaksız bir şekilde devam edemezsin. Ben bu ameliyatı yaklaşık bir yaşımdayken oldum. Çeneyi kırıyorlar ve damağa dikiş atıyorlar. Ya da vücudundan bir parça alıp damağa ekleme yapıyorlar. Bu durum tabii ki ağızda bozulmaya da yol açıyor. Mesela benim damağımda iki tane daha dişim var, buna bağlı olduğunu düşünüyorum. O dişlerimi de çekemiyorum çünkü damağımın tekrardan düşme riski var.
Dudak damak yarıklığı hastalığı için kurulan dernekleri yeterli buluyor musunuz? Derneklerden ihtiyacın olan manevi desteği alıyor musunuz?
Böyle bir dernek olduğunu bilmiyordum. Az önce baktım, varmış. Hiç haberim yoktu gerçekten. Aslında bu biraz daha artabilir çünkü dediğin gibi, maddiden ziyade -tabii maddi durumu kötü olan aileler ameliyat masraflarını karşılayamayacaktır ama- manevi desteğin biraz daha fazla olmasından yanayım çünkü benim görünüşüme bakınca herhangi bir aksaklık görünmüyor, dudağıma ya da burnuma atlamamış. Ama bu hastalıkta burnuna kadar eksiklik olan -delik şekilde- insanlar var ve bu toplum tarafından garipseniyor. Dış görünüş olarak çok farklılar ve özellikle bu konuda insanlara psikolojik destek sağlanması gerekiyor.
Hastalığınızda tedaviye, rehabilitasyona ve bakım hizmetine ulaşmada zorluk yaşadınız mı?
O zamanlar Doğu’da bir şehirde yaşıyorduk -hastalığımın fark edildiği zaman- ve orada benim terapi alabileceğim bir yer yoktu. Damak olmadığı için konuşmak daha çok burundan ve genizden oluyor. Birçok harfi çıkartmada zorluk yaşanıyor. Hala arada sırada zorlandığım anlar oluyor fakat çocukken söyleyemediğim çok harf vardı. Bunun için konuşma terapisi almanız gerekiyor. Sırf tedavi almam gerektiği için Ankara’ya taşınmıştık. İki yıl kadar konuşma terapisi aldım, dört yaşımdan altı yaşıma kadar. Ama yine de alınan terapinin yüzde yüz etkisi olmuyor. Ben birçok hastaya göre konuşma konusunda daha iyiyim aslında, çok daha kötü seviyelerde olanlar da var.
Nadir hastalığın en önemli psikososyal etkilerinden biri de izolasyondur. Nadir hastalığı bulunan kişiler ve aileleri, hoş olmayan ya da utandırıcı durumlar yaşama ihtimali nedeniyle bazı sosyal durumlardan kaçınmaktadır. Dudak damak yarıklığı hastalığı sosyal hayatınızı etkiliyor mu?
Aslında normalde etkilemesi gerekli. Bunlar kişiden kişiye göre değişir tabii ki ama beni hiç etkilemedi. Bu durum benim öz güvenimi hiç kırmadı. Terapi aldım, konuşmam çok iyi bir noktaya geldi fakat hala bazı harfleri çıkartmakta zorluk çekiyorum. Dudak ünsüzlerini çıkartmakta zorlanıyorum mesela. Ya da balon şişiremiyorum örneğin, bu çok küçük bir örnek olacak ama… Tabii insanlar anlıyor bir aksaklık olduğunu fakat şimdiye kadar pek de umrumda olmadı açıkçası. Hatta her zaman en çok konuşan insan ben oldum.
-Nadir hastalığı bulunan kişi ve ailelerin yarısından fazlasının anksiyete ve korku, yarısına yakınının ise öfke, hayal kırıklığı ve belirsizlik duyguları yaşamakta olduğu ortaya konulmuştur. (pelentsov vd., 2016, s. 24)- Anksiyete, korku, öfke, hayal kırıklığı ve belirsizlik gibi duygular yaşıyor musunuz? Bunun için tedavi alıyor musunuz ya da almak ister misiniz?
Aslında çok fazla anksiyete yaşadığım olmuyor ama ister istemez bazı anlarda oluyor. Bakıldığında hayatın en temel unsuru, ya da sosyal hayatın en temel unsuru diyebiliriz konuşmak için. Birçok insan da hastalığımı fark etmiyor doğrusu, genelde ‘geniz etin mi var?’ sorusunu duyuyorum ama aslında yok. Mesela okulda, ilk tanışma zamanlarında, insanları tanımadığım için gerginlik yaşıyordum. Bazı anlarda insanların garip bakışlarına maruz kalabiliyorum ama dediğim gibi benim durumum öyle çok üst düzey değil, tabii öyle olan insanlar vardır bu kişiden kişiye göre değişir. Tedavi almıyorum, anksiyetemin o boyutta olduğunu düşünmüyorum.
‘ BİR COĞRAFYADA SIK GÖRÜLEN HASTALIKLAR SONRADAN SONRAYA NADİR HASTALIKLAR GRUBUNA DAHİL OLABİLİR’
Enfeksiyon hastalıkları ve klinik mikrobiyoloji uzmanısınız, branşınızla ilgili olan nadir hastalıkların sayısı fazla mı?
Bizim branşımızla ilgili nadir hastalıklar çok sayılabilir. Çünkü büyük bir mikrobiyolojik dünyanın üyelerini içeriyor. Tasnif edecek olursak bakteriler, virüsler, mantarlar, parazitler… enfeksiyon hastalıkları mikrobiyota dediğimiz çok geniş bir evrene ait mikroorganizmalara bağlı olarak meydana gelen hastalıklardır. Bu hastalıkların hepsi her toplumda aynı sıklıkta görülmez. Dolayısıyla bir kısmı oldukça nadir görülür, bir kısmı ise bazı toplumlarda çok yüksek oranda görülür. Bu sıklıkla görülme işi enfeksiyon hastalıklarında bazı kavramlarla ifade edilir. Örneğin; endemi kavramı. Endemi demek belirli bir hastalığın, belirli bir toplumda sürekli olarak bulunma sıklığı demektir. Son zamanlarda sıklıkla duyduğunuz kelime: pandemi. Pandemi, bir hastalığın kıtalar arası yayılımı demektir. Bütün dünyayı ilgilendiren, salgın olarak nitelendirebileceğimiz hastalıkları pandemi kavramı altında toplarız. Bir de Sporadik dediğimiz kavram vardır. Sporadik, belirli coğrafi alanlarda, belirli zamanlarda ortaya çıkan nadir hastalıklardır. Epidemi, belirli coğrafi alandaki salgındır. Örneğin, sadece Türkiye’yi ilgilendiren bir salgın ortaya çıktığında buna epidemi deriz. Ama bu salgın, Türkiye’nin sınırlarını aşıp kıtalar arasında yayılmaya başladığında pandemi adını veririz. Biz hastalıklarımızın sıklıklarıyla ilgili bu tür kavramlar kullanıyoruz.
Enfeksiyon hastalıkları ve klinik mikrobiyoloji ile ilgili birkaç nadir hastalık örneği verebilir misiniz?
Genellikle -kendi yaşadığımız coğrafya Türkiye için söyleyecek olursak- daha çok tropikal hastalıklar nadir hastalıklar grubundadır. Özellikle paraziter hastalıklardan örneğin sıtma. Eskiden daha sık görülüyordu ama şimdi oldukça nadir görülmektedir. Yine paraziter hastalık olarak sayabileceğimiz trypanosoma bizde çok nadir olarak görülen hastalıklardır. Viral hastalıklar içerisinde örneğin ebola virüsüne bağlı olan hastalıklar bizde çok nadir görülür. Örneğin AIDS virüsü olan HIV Afrika toplumunda ve gelişmiş bazı ülkelerde çok sık görülmesine rağmen hala ülkemizde nadir sayıda görüldüğünü söyleyebiliriz. Birçok mantar enfeksiyonları da aynı şekilde bizim coğrafyamızda çok nadir şekilde gördüğümüz hastalıklardır. Belirli mikroorganizmaları çok sık görürüz ama belirli mikroorganizmaları daha az olarak görürüz. Özellikle mantar enfeksiyonları bağışıklık sistemi düşmüş olan, kanser kemoterapisi gören, organ transplantasyonu yapılmış olan kimselerde daha sıklıkla görüldüğü için diğer genel olarak diğer enfeksiyon hastalıklarıyla kıyasladığımızda çok nadir görülen hastalıklardır. Bunların çeşitli isimleri var tabii ama tıbbi terminoloji olduğu için çok detaya girmek istemem.
Branşınızla ilgili bir nadir hastalığa yoğunlaşalım ve üzerinde konuşalım. Yoğunlaşmamızı tercih ettiğiniz bir hastalık var mı?
Mesela eskiden Türkiye’de daha sıklıkla görülen sıtma konusunu ele alabiliriz. Ve buna gelmeden önce enfeksiyon hastalıklarını günümüzde iki ana kategoriye ayırmamız mümkün. Bunlardan bir tanesi toplumdan kazanılan enfeksiyon hastalıkları yani toplumda dolaşımda bulunan mikroorganizmalardan insanlara bulaşan hastalıklar. Bir diğer kategorimiz ise güncel olarak özellikle enfeksiyon hastalıkları uzmanlarını daha sıklıkla ilgilendiren kategori ise sağlık hizmetleriyle ilişkili enfeksiyonlardır. Teknolojik gelişimle beraber insan yaşamının kalitesinin artması, ömrünün uzaması ancak bununla beraber insanlarda kanser olgularının çok fazla artmış olması, organ transplantasyonunun artmış olması, immün sistemi baskılayacak tedavilerin uygulanmış olması, çok sayıda kronik rahatsızlığı. benzer nitelikteki hastalar sağlık tesislerinde kümelendiğinde ortaya çıkan mikroorganizmalar, çok sayıda kullanılan antimikrobiyallere karşı direnç geliştirmekte ve tedavisi güçlük oluşturmaktadır. Böyle olunca bu mikroorganizmalar günümüzde ciddi sorunlar oluşturmaktadır. Hem maliyet açısından, hem hastanede kalma sürelerinin uzaması açısından, hem de hastaların yaşam kalitelerini düşürmesi bakımından son derece sıkıntılara yol açabilmektedir. Örneğin, daha öncelerde diziyle ilgili bir problemi olan bir kişinin dizi belirli bir seviyeden kesiliyordu ama şimdi protez adını verdiğimiz, dışarıdan yapılmış, özel nitelikli, vücuda uyumlu metallerin vücut içerisine yerleştirilmesi söz konusu. Ancak, bu cansız bir yapı olduğundan dolayı vücutla uyumu bazen olamamakta veyahut da ameliyat sırasında o bölgeye mikroorganizma -hastaneden- bulaşacak olursa o kişinin proteziyle ilgili çok uzun süre enfeksiyon gelişmesine, yaşam kalitesinin bozulmasına hatta gerekirse protezin çıkarılmasına sebebiyet verebilmektedir.
Nadir görülen enfeksiyon hastalığı olarak ülkemizde daha önce özellikle akdeniz bölgesi, güneydoğu anadolu bölgesinde ve doğu anadolu bölgesinde daha tropikal iklime yakın bölgelerde daha çok görülen ama günümüzde Sıtma Savaş ile yapılan mücadele neticesinde artık çok nadir görülen sıtma konusuna değinebiliriz.
Sıtma hastalığı bulaşıcı mıdır?
Sıtma, plasmodium adını verdiğimiz bir parazit tarafından bulaşmaktadır. Bu bulaşma doğrudan doğruya insana bulaşmıyor ancak dişi anofel dediğimiz sivrisinek cinsi, hasta bir kişinin kanını emerek kendi bağırsaklarında bu plasmodiumun olgunlaşma evresini sağlıyor ve belirli bir aşamaya geldikten sonra o dişi sivrisinek bir başka kişiyi ısırdığında o paraziti kana veriyor. Parazit kana karıştıktan sonra özellikle kandaki alyuvarlar içerisine yerleşebiliyor ve kan organları içerisine (karaciğer, dalak vs.) yerleşerek bunlarda büyümeye, patolojiye yol açıyor. Ciddi bir çoğalma evresinden sonra da insanlarda çok ciddi hastalıklara, hatta belirli türleri ölüme kadar sebebiyet verebiliyor. Bu enfeksiyon hastalıklarının genelde belirtileri çok spesifik olmayabilir. Yani, kişilerdeki bazı rahatsızlıklar insanlarda bu hastalık şudur dedirtmez. Genellikle yüksek ateş, üşüme titreme, öksürük, karın ağrısı, ishal, sık idrara çıkma, baş ağrısı, yaygın vücut ağrısı, kas ağrısı, terleme gibi belirli belirti ve bulgular birçok enfeksiyon hastalığında görülebilir. Dolayısıyla, nadir hastalıkların enfeksiyon hastalıkları uzmanlığı yönünden dikkat edilmesi gereken şey yüksek olasılıkla aklınıza gelmesi diyebiliriz. O yüzden enfeksiyon hastalıklarında tecrübe son derece önemli. Toplumdan edinilen enfeksiyonların çok sıklıkla karşılaşıldığı yerlerde çalışmış olmak -özellikle Afrika’da, Güney Amerika’da, Kuzey Amerika’da, Orta Doğu’da-, o bölgelerde karşılaşılan enfeksiyonları görmek ve onun dışında hastanelerde, yoğun bakımlarda yatan hastalarla ilgili enfeksiyonları görmek tecrübe bakımından son derece önemli.
Sıtma hastalığı nasıl teşhis edilir? En yaygın belirtileri nelerdir? Tedavisinde kullanılan bir ilaç var mı?
Sıtmanın belirtileri başlangıç için son derece non-specifictir, tanı koydurmayabilir ancak enfeksiyon hastalıkları uzmanı bu şahsın rahatsızlığının ne zaman başladığı, nerede yaşadığı, yaşam koşulları, bulunduğu bölgenin iklim özellikleri, bu hastalığın endemik olduğu bölgelere seyahatinin olup olmadığı gibi epidemiyolojik soruları sorarak onunla ilgili adeta bir dedektif gibi belirli bir şeye ulaşabilir, şüphelenebilir. Şüphelendikten sonra hastayla ilgili yapılması gereken bazı tahliller vardır. Örneğin sıtma için, hastanın parmak ucundan alınacak, özellikle ateşin yükselmeye başladığı üşüme, titreme ve ateş periyodunda alınacak bir damla kanın bir lam üzerine alınması ve ona özgü, spesifik boyalarla boyadıktan sonra mikroskop altında incelenmesiyle tanı, doğrudan konulabilmekte. Sıtma gibi toplum sağlığını ilgilendiren bu tür rahatsızlıklar için ülkemizde de -birçok ülkede olduğu gibi- mücadele birimleri kurulmuştur. Örneğin verem mikrobu için Verem Savaş birimleri kurulmuştur. Sıtma ile ilgili ise Sıtma Savaş birimleri… ancak sıtma çok nadir görüldüğü için artık bizim için endemik kategorisinde sayılmayabilir. Günümüzde bulaşıcı hastalıklar kategorisinde takip ediliyor. Toplum sağlığını ilgilendiren hastalıkların tedavisinde genellikle ülkelerin bakanlıkları bu görülen parazite yönelik olarak hangi cinsi, hangi tipi görülüyorsa; onları yok edebilecek, onlara karşı etkili olabilecek ilaçları bünyesinde bulundurmaktadır. Toplum sağlığını ilgilendirdiği için belirli bir kayıt sisteminde takip edilmektedir. yani, bir kişi sıtma olduğu zaman, herhangi bir özel ya da kamu hastanesinde tıbbi hizmet alabilir ama toplum sağlını ilgilendirdiği için ister özel kuruluşa başvurmuş olsun ister kamu kuruluşuna başvurmuş olsun bu şahsın kaydı mutlaka sağlık bakanlığının ilgili birimlerine bildirilir. Dolayısıyla bakanlık hangi bölgede ne kadar vaka olduğunu bilir ve bir salgın tehditi halini almadan hemen önlemleri alabilmesi için bu son derece gerekli bir durum. Buna yönelik ilaçları bakanlık kendi bünyesinde bulundurur. Böyle bir hasta olduğu zaman, o hastaya enfeksiyon hastalıkları uzmanının takibinde ilaçları kullanması ve akabinde de kontrolü sağlanarak tam şifaya kavuşana dek takibi yapılmış olur.
Sıtma hastalığı yaşa ve cinsiyete göre değişkenlik gösterir mi?
Tabii. Özellikle çocukluk çağında farklılık gösterebilir. Örneğin, bir hastamıza Hindistan’da böbrek transplantasyonu yapılmıştı. Böbrek transplantasyonu sonrasında vücudun yabancı olan böbreği atmaması için ona kendi immün sistemini baskılayacak ilaçlar verilir. Dolayısıyla immün sistemi baskılayacak ilaçlar verildiği için hastada belirli bir mikrobik rahatsızlığın semptomlarının ortaya çıkmasını baskılar. Örneğin, ateşinin çıkmasını önler. Her kişide her enfeksiyon hastalığının belirtisi aynı olmayabilir. Çocuklarda farklı seyredebilir, gebelerde farklı seyredebilir, yaşlılarda farklı seyredebilir, kronik alt hastalığı olanlarda farklı seyredebilir, kanser hastalarında farklı seyredebilir.
Sıtma hastalığının kaç alt tipi vardır?
Sıtmayı oluşturan etkenler değişir. Mesela plasmodium dediğimiz parazitin genel bir ismidir. Plasmodium vivax, plasmodium malariae, plasmodium ovale, plasmodium falciparum şeklinde species dediğimiz türleri vardır. Bunların oluşturduğu klinik tablolar da birbirinden farklıdır. Bizde en sıklıkla görülen plasmodium ovaledir, orta şiddette görülen bir rahatsızlığa yol açar. Özellikle Afrika’da bazı bölgelerde Nijerya, Mozambik gibi yerlerde plasmodium falciparum cinsi görülmektedir. Falciparum cinsinin halk arasında karasu humması olarak bilinir. Sebebi çok ölümcül seyretmesidir ve alyuvarları parçaladığı için insanların adeta idrarlarında yalnızca kan varmışcasına bir görünüme yol açar. Nörolojik tutulum çok fazla olduğu için ölümle seyreden bir formdur. Alt grupları öldüren ilaçlar da birbirinden farklıdır.
Doğumla beraber gelen ve Genetik Hastalıklar; Kalp Hastalıkları 4
Alt Tipler
Dilate kardiyomiyopati-1S
Hastalığın Grubu
Dilate kardiyomiyopati
Dilate kardiyomiyopati (DCM) – basitçe kalp yetmezliğine ve ritim bozukluğuna (aritmi) sebep olan- ventriküler dilatasyon ve bozulmuş sistolik fonksiyon ile ilişkilendirilmiş karakterize bir kalp kası hastalığıdır.
Herhangi bilinen bir neden olmadan bir ailenin herhangi bir üyesinde görüldüğünde hastalık idiyopatik olarak adlandırılırken, yine herhangi belirli bir sebep olmadan aynı ailenin bireylerinde görülmesi durumuna ailevi izole dilate kardiyomiyopati adı verilir. 3Karşılaşılan vakaların %20-48’i kalıtsal olup ailevi dilate kardiyomiyopati (FDC) olarak da adlandırılır. Kardiyak miyositteki sitoskeletal ve sarkomerik proteinleri kodlayan FDC genlerinde görülen bir mutasyon hastalığın ortaya çıkmasında başlıca etkendir.3
Genellikle yirmili ve otuzlu yaş dönemlerinde ventriküler dilatasyon ve sistolik disfonksiyon gelişimi ile kendini gösterse de5 hastalık her yaşta ve her cinsiyette görülebilmektedir. 1
Kalıtım Paterni/Deseni
Ailevi dilate kardiyomiyopati, yaşa bağlı penetrans (Bir genin fenotipte görülme olasılığı) ile otozomal dominant kalıtım deseni ile karakterizedir 5veilgili gene göre değişiklik gösteren farklı kalıtım paternlerine/ desenlerine sahiptir. Bu hastalığın %80 ila 90’ı otozomal dominant model ile taşınırken, nadir durumlarda (%10-20’si) otozomal resesif genler ile taşındığı gözlemlenmiştir. Bunların yanı sıra, bazı nadir vakalarda hastalığın X kromozomu ile taşınabildiği de gözlemler arasındadır.2 Bazı çalışmalarda Mitokondriyal kalıtım ile iletildiği de görülmüştür.1
Hastalık her türlü kalıtım yoluyla iletildiği için, soy geçmişi taraması yaparaktan ailevi kaynaklı olup olmadığı teşhis edilebilir. Bununla beraber, teşhisi kolaylaştırmak için bazı FDC genlerine özgü klinik testler geliştirilmiştir.
Hasta yakınlarının kapsamlı taraması da bu hastalığın standartlarındandır. Elektrokardiyogram (EKG) ve ekokardiyografi kullanılarak özellikle birincil akrabaların klinik taraması, FDC’nin tanımlanmasına müsaade eder. Bunlara ek olarak Sinyal ortalamalı elektrokardiyografi (SAECG) de bir tanı aracı olarak önerilmiştir. Serum kreatinin kinaz seviyeleri, özellikle bu hastalığın incelenmesine sebep olan kişide (proband) artışa dair işaret görülürse, diğer aile bireylerinde uygulaması faydalı olabilir.
Ayrıca, genetik danışmanlık yöntemi de kullanılarak belirti geliştirme riski taşıyan aile bireyleri belirlenir. Lakin genetik danışmanlık yöntemi uygulanırken yaşa bağlı sorunlar ve genotip ve fenotip arasındaki eksik geçişler (inkomplent penetrans) göz önünde bulundurulmalıdır. Bu tür teknikler, hastalığın erken teşhisinde yahut belirti göstermeden önce teşhis edilmesinde önemli rol oynamaktadır. Erken teşhis her açıdan kolaylık sağlar.
Hastalığın Yönetimi ve Tedavisi
FDC’nin yönetiminde kalp yetmezliğinin ilerlemesini sınırlandırma ve ritim bozukluğunu (aritmi) kontrol etme başı çekerken DCM için de uygulanan genel tedavi yöntemleriyle beraber, ilaç tedavileri (farmakoterapi) ile destek cihazları da kullanılmaktadır.
Genel Önlemler
Hastanın hastalığı konusunda eğitilmesi
Tuz ve sıvı kısıtlaması
Hipertansiyon tedavisi
Alkol alımının kısıtlanması
Vücut ağırlığının kontrolü
Orta düzeyde egzersizler – kontrollü bir ortamda
İlaç Tedavisi (Farmakoterapi) – Belli başlı ilaç tedavilerinden ve detaylarından bahsedilmiştir.
CONSENSUS, SOLVD ve SAVE dâhil olmak üzere birçok çalışmada belirtildiği gibi, kalp yetmezliğinin ilerlemesini, hastaneye yatış oranları ile beraber ölüm oranlarını (mortalite) azaltmakta etkilidir.
Bu tür ilaçlarda düşük dozda başlanır ve randomize çalışmalarda etkinlik gösterenlerde kademeli bir şekilde artırılarak ilaç ayarlaması yapılır.Kaptopril günde üç defa olmak üzere en fazla 50 mg’a çıkarılır. Enalapril için maksimum düzey günde iki defa olmak üzere 20 mg iken Lisinopril için günde bir kez 40 mg’dır.
Anjiyotensin Reseptör Blokerleri (ARB’ler)
ACE inhibitörlerine toleranssız hastalar için uygun bir alternatiftir. ARB’nin kullanımı ELITE-I ve II, Val-HeFT ve OPTIMAAL gibi denemelere dayanmaktadır. Bu çalışmalarda günde bir defa yaklaşık 50 mg Losartan kullanıldı. Valsartan için en yüksek miktar günde iki defa 160 mg’dır. Val-HeFT denemesine göre, bir ARB’nin bir ACE inhibitörüne eklenmesinin fazladan yarar sağlama ihtimali çok düşüktür.
Birinci Nesil Kalsiyum Kanal Blokerleri
Kalp yetmezliği olan hastalara önerilmez.
Endotelin Antagonistleri
Denemeler bugüne kadar hayal kırkılığı ile sonuçlanmıştır ve standart kılavuzlarda önerilmemektedir.
Diüretikler (idrar söktürücü ilaçlar)
Kalp yetmezliğinde hayatta kalma üzerindeki etkilerini doğrulamak için herhangi bir randomize çalışma yapılmamıştır. Furosemide’nin günlük kullanım miktarı 20 mg ila 600 mg’dır. Bumetanide ve ekatrinik asit şu anda kullanımda olan diğer loop (döngü) diüretiklerdir. Torsemid ağız yoluyla alındığında daha etkilidir ve oral furosemide yanıt vermeyen bazı hastalarda kullanılır. Çoğunlukla, benzer sonuçlar için furosemide’nin yarı dozu etkilidir. Yüksek doz döngü diüretiklerine ihtiyaç duyulduğunda ve özellikle hastanın diüretik direnci varsa, genellikle metolazon günde bir kez 2.5 mg ila günde iki kez 5 mg miktarında eklenir. Asetazolamid, metabolik alkalozlu bazı hastalarda günde 250-500 mg miktarında kullanılmaktadır.
Aldosteron İnhibitörleri
Ayrı ayrı RALES VE EPHESUS’da belirtildiği gibi, Spironolakton ve Eplerenon gibi aldosteron inhibitörleri ölüm oranlarını azaltır.
Spironolakton, günde bir defa 12,5 ila 50 mg arasında değişen dozlarda kullanılır. Eplerenon, ağız yoluyla alınarak, günde 25 mg’lık dozda başlanır ve hasta toleransına göre bu miktar günde bir defa 50 mg’a çıkarılabilir.
Eplerenon aldosteron reseptörleri için daha çok tercih edilir ve jinekomasti gibi spironolaktonla ilişkili bazı yan etkileri önler.
Vazopressin Antagonistleri
Hala araştırılmaktadır.
Natriüretik peptitler
Yalnızca akut dekompansasyon için doğrudan damar yoluyla verilebilmektedir(intravenöz).
Digoksin
Kronik kalp yetmezliği tedavisinde yaygın olarak kabul edilen tek inotroptur.Dozaj ayarlaması böbrek fonksiyonlarına göre 0.125-0.25 mg olarak ayarlanır ve günde bir kez uygulanır. Digitalis Investogators Group’un [32] yakın tarihli bir post-hoc analizi hastaları digoksin serum seviyelerine göre gruplandırmış ve 0,5 ile 0,8 ng / ml düzeyinde seyreden hastaların alt grubunda ölüm oranlarının sebepleri fark etmeksizin %6.3 oranında azaldığı gözlemlemiştir.
Beta-Adrenerjik Blokerler
Kalp yetmezliği tedavisinde ciddi ilerlemeler kaydettiği görüşü hâkimdir. CIBIS-II [33] ve MERIT-HF [34], bisoprolol ve metoprolol süksinat (her ikisi de beta-1 seçici bloker) kullanıldığında nedeni ne olursa olsun ölüm oranlarında %34’lük bir oranda azalma gözlemlenmiştir.
Alfa bloke edici özelliklere sahip seçici olmayan bir beta bloker olan Carvedilol’ün sayesinde ağır derecede kalp yetmezliğinden (COPERNICUS) kaynaklanan ölümlerin %35 azaldığı gözlemlenmiştir.
Antikoagülanlar / Antiplatelet Ajanlar
Hala kullanılması tartışmalı olan ve henüz kim için ve hangi tedavi için kullanılması hakkında yeterli çalışma olmayan ilaç tedavi yöntemlerindendir. Klinik çalışmalarda genellikle sol ventriküler ejeksiyon fraksiyonu% 30’dan az olan hastalarda kullanılmaktadır.3
Hastalığın şu ana kadar bilinen ve HPO –İnsan Fenotip Ontolojisi- tarafından yayımlanan tabloda görüldüğü üzere on dört belirtisi bulunmaktadır. Kişiden kişiye değişen belirtilerden dolayı bu sayı her seferinde artmakta ve tablo güncellenmektedir.
Tıbbi Terimler
Diğer İsimler
Görülme Sıklığı
Sol ventrikül hipertrofisi
–
%80-%90
Atriyal fibrilasyon
Düzensiz kalp atışına neden olan üst kalp odacıklarının titremesi
%30-%79
Konjestif kalp yetmezliği
Kalp yetmezliği, kardiyak yetmezlikler, kalp yetmezliği
FDC’NİN yayılım ve görülme sıklığı tam olarak bilinmemekle birlikte dilate kardiyomiyopati’nin görülme sıklığı dünya çapında 1/12,000-28,000 arasında tahmin edilirken yaygınlığı tahminen 1/2500’dir. Ailevi izole dilate kardiyomiyopati (FDC) dilate kardiyomiyopati vakalarının yaklaşık %20-30 (% 2-65 aralığı) ‘unu oluşturmaktadır. 1
OMIM.org’da bildirildiğine göre, yaygınlığı 100.000’de yaklaşık 36.5’tir. ABD’de yılda 10.000’den fazla ölüme neden olur ve kalp nakli için tıbbi gerekliliği (endikasyon) birincildir. Distrofin’de oluşan mutasyon nadir durumlar ((örneğin, 300377.0021) dışında, ailesel oluşum 20 ila% 25’tir.5
FDC’nin %30 ila 40’ı elliden fazla gene kaynak gösterilerek tanımlanmıştır. Ancak bu sayı sabit değildir, yeni tanılar bu sayıyı değiştirmektedir.
Mutasyonların çoğu titin (TTN;% 15-27), lamin A / C (LMNA;% 6), Beta-miyozin ağır zinciri (MYH7;% 4.20), miyopalladin (MYPN;% 3.50) ve kardiyak troponin T (TNNC1, TNNI3 ve TNNT2;% 2.90) proteinlerinde görülmüştür.1
Şu ana kadar üzerinde inceleme yapılmış FDC genleri: DES, DMD, LMNA, MYBPC3, MYH7, TAZ, TNNT2 ve TMP1. 3
Prognoz (Hastalığın sonucunu tahmin)
Hastalığın sonucu hakkındaki tahminler hastalığın herhangi bir mutasyona uğraması ile değişebilir, ancak tıbbi olan veya olmayan tedaviler ile ölüm oranları azaltılıp sonuç hakkında tahminlerde bulunulabilir. 1
Karnitin uzun zincirli yağ asitlerinin mitokondri içerisine taşınmasına yardım eder. Karnitin palmitoil transferaz I (CPTI) uzun zincirli yağ asit moleküllerini karnitine bağlar. Karnitin Açilkarnitin Translokaz (CACT) oluşan birleşik molekülün mitokondrial iç membrandan geçmesini sağlar. CPT II ise birleşiği karnitinden ayıklayarak çözer. Yeterince işleyen karnitin palmitoiltransferaz 2 olmadan, uzun zincirli yağ asitleri mitokondriye girdikten sonra uygun şekilde işlenmez ve enerji üretmek için metabolize edilemez. Azalan enerji üretimi, CPT II eksikliğinin kas ağrısı ve zayıflığı, düşük kan şekeri (hipoglisemi) ve yağ parçalanması ürünlerinin düşük seviyeleri (hipoketoz) gibi bazı özelliklerine yol açabilir. Yağ asitleri ve uzun zincirli açilkarnitinler (hala karnitine bağlı olan yağ asitleri) hücrelerde birikebilir ve karaciğere, kalbe ve kaslara zarar verebilir. Bu anormal birikim, bozukluğun diğer belirti ve semptomlarına neden olur. Otozomal resesif bir hasalıktır.
Üç ana tip CPT II eksikliği vardır: Ölümcül bir yenidoğan formu, şiddetli bir infantil hepatokardiyomüsküler form ve bir miyopatik form. CPT II eksikliğinin ölümcül neonatal formu doğumdan hemen sonra ortaya çıkar. Bu bozukluğa sahip bebeklerde solunum yetmezliği, nöbetler, karaciğer yetmezliği, zayıflamış kalp kası (kardiyomiyopati) ve düzensiz kalp atışı (aritmi) gelişir. Etkilenen kişilerde ayrıca düşük kan şekeri (hipoglisemi) ve yağların parçalanması sırasında üretilen ve enerji için kullanılan düşük keton seviyesi vardır. Bu belirtilere birlikte hipoketotik hipoglisemi denir. Çoğu durumda, beyin ve böbrekler de yapısal olarak anormaldir. CPT II eksikliğinin ölümcül neonatal formu olan bebekler genellikle birkaç gün ila birkaç ay yaşarlar. CPT II eksikliğinin şiddetli infantil hepatokardiyomüsküler formu karaciğeri, kalbi ve kasları etkiler. Belirtiler ve semptomlar genellikle yaşamın ilk yılında ortaya çıkar. Bu form, tekrarlayan hipoketotik hipoglisemi, nöbetler, genişlemiş bir karaciğer (hepatomegali), kardiyomiyopati ve aritmi ataklarını içerir. Bu tür CPT II eksikliği ile ilgili sorunlar, açlık dönemleri veya viral enfeksiyonlar gibi hastalıklar tarafından tetiklenebilir. CPT II eksikliğinin şiddetli infantil hepatokardiyomüsküler formu olan kişiler, karaciğer yetmezliği, sinir sistemi hasarı, koma ve ani ölüm riski altındadır. Miyopatik form, CPT II eksikliğinin en az şiddetli türüdür. Bu form, tekrarlayan kas ağrısı (miyalji) ve güçsüzlük atakları ile karakterizedir ve kas dokusunun bozulması (rabdomiyoliz) ile ilişkilidir. Kas dokusunun tahrip edilmesi, böbrekler tarafından işlenen ve idrarda (miyoglobinüri) salınan miyoglobin adı verilen bir proteini serbest bırakır. Miyoglobin, idrarın kırmızı veya kahverengi olmasına neden olur. Bu protein ayrıca böbreklere de zarar verebilir ve bazı durumlarda yaşamı tehdit eden böbrek yetmezliğine yol açabilir. Miyalji ve rabdomiyoliz epizotları egzersiz, stres, aşırı sıcaklığa maruz kalma, enfeksiyonlar veya oruç tutma ile tetiklenebilir. İlk bölüm genellikle çocukluk veya ergenlik döneminde ortaya çıkar. Miyopatik CPT II eksikliğine sahip çoğu insan, bölümler arasındaki bozukluğun hiçbir belirti veya semptomuna sahip değildir.
Hastalıkla İlişkili Genler
Karnitin palmitoil transferaz I (CPTI)
Karnitin palmitoil transferaz II (CPTII)
Karnitin Acilkarnitin Translokaz
Teşhis Yöntemleri ve Tedaviler
Teşhis, serum/plazma açilkarnitinlerin ilk tandem kütle spektrometrisi ve ardından, taze dolaşımdaki lenfositler, kaslar veya fibroblastlarda mutasyon analizi ve CPT2 enzim aktivitesinin ölçümleri ile yapılır. Tanı açilkarnitin profili ile konulabilir. Tedavi için genel olarak Bezafibratı hafif CPT2 eksikliği olan 6 yetişkine 6 ay süreyle günde 3200 mg tablet dozunda uygulanmıştır. Aynı zamanda bu tür tedaviler, uzun süreli açlıktan ( > 12 saat) kaçınmaya ve düşük yağlı ve yüksek karbonhidratlı diyete dayanır.
Hastalığın Diğer İsimleri
Carnitine palmitoyltransferase deficiency type 2
CPT2
Carnitine palmitoyltransferase II (CPT II) deficiency
Sporadik Creutzfeldt-Jakob hastalığı, İngilizce adı ile sporadic Creutzfeldt-Jakob disease (sCJD) beyindeki anormal prion proteinlerinin birikmesinden kaynaklanır. Çoğu hasta için anormal prionların nedeni bilinmemektedir (sporadik CJD). sCJD, CJD’nin %85’ini oluşturur ve prion protein yapısındaki rastgele (sporadik) değişikliklerden kaynaklandığı düşünülmektedir. Bazı araştırmacılar, normal hücresel prion proteininin (PrPc), beyin hücrelerinin dejenerasyonunu ve kaybını önlemede önemli bir rol oynadığını düşünmektedirler. CJD’nin diğer nedenleri %10-15 genetik ve %1 edinseldir.
Hastalığa neden olan değiştirilmiş veya “yanlış katlanmış” PrPc formu, PrPsc olarak bilinir. PrPsc, normal PrPc’nin hastalığa neden olan forma dönüşmesine neden olabilir. PrPsc’nin yanlış katlanmış şeklinin vücut tarafından uygun şekilde parçalanmasını engellediği düşünülmektedir. Sonuç olarak, giderek daha fazla normal PrPc, yavaş yavaş birikerek beyinde sabit tortular (plaklar) oluşturan PrPsc’ye dönüşür
Belirti ve Semptomlar
sCJD ‘de semptomlar tipik olarak 40-60 yaşları arasında ortaya çıkar. Başlangıçta bu belirtiler kafa karışıklığı, depresyon, unutkanlık, uyku güçlükleri (uykusuzluk) ve/veya davranış değişiklikleri olarak görülür. Etkilenen bireyler ayrıca görme bozukluğu, anormal fiziksel his ve/veya istemli hareket koordinasyonunda zorluklar yaşayabilir.
Bireyler daha sonra hızla ilerleyen entelektüel yetenek kaybı yaşayabilir, bozulmuş hafıza ve muhakeme yeteneği ve belirgin kişilik değişiklikleri (demans) gösterebilir. Nöromüsküler anormallikler, bozukluğun bu aşamasında daha belirgin hale gelir ve kas zayıflığı ve kas kütlesi kaybı olabilir. Kas sertliği, titreme, özellikle kol ve bacaklarda tekrarlayan, istemsiz, şok benzeri kas spazmları (miyoklonus) ve/veya yavaş, sürekli, istemsiz hareketler(atetoz) görülebilir. Gönüllü hareketlerin giderek artan şekilde bozulmuş koordinasyonu ve/veya bozulmuş kas kontrolü nedeniyle konuşma güçlüğü (dizartri) gelişebilir. Görme de giderek bozulabilir.
sCJD’li bireylerde, nörolojik ve nöromüsküler bozukluk ilerlemeye devam eder ve bozukluğun sonraki aşamaları, fiziksel ve entelektüel işlevlerin kaybı, koma ve solunum yollarının tekrarlayan enfeksiyonlarına (örn., pnömoni ( zatürre)) karşı artan duyarlılık ile karakterize olur. Birçok hastada, yaşamı tehdit eden komplikasyonlar, hastalık belirginleştikten bir yıldan daha kısa bir süre içinde gelişmektedir.
Teşhis Yöntemleri
sCJD’yi teşhis etmek çok nadir bir hastalık olması ve diğer demans formlarına benzemesi dolayısı ile çok zordur. Teşhisi doğrulamanın tek ve kesin yolu, beyin biyopsisi veya otopsi ile yapılabilecek küçük bir beyin dokusu örneğini test etmektir.
Klinik semptomların yanı sıra, olası bir CJD şüphesini belirlemede faydalı olabilecek diğer testler de vardır. İlki, gerçek zamanlı sarsıntı kaynaklı dönüşüm (RT-QuIC) olarak adlandırılır. Bu yöntem, omurilikten gelen sıvıda prion birikimlerinin oluşumu yoluyla anormal prion proteini arar. İkincisi, manyetik rezonans görüntüleme (MRI) ile beyin görüntülemedir. Uzmanlar, RT-QuIC, MRI ve klinik semptomlardan elde edilen sonuçları kullanarak, bir bireyin olası bir CJD teşhisi için kriterleri karşılayıp karşılamadığını belirleyebilir. Göz önünde bulundurulabilecek diğer bir test de beynin elektriksel uyarılarını kaydeden elektroensefalogramı (EEG) ‘dır.
Tedaviler
Hastalığın kesin bir tedavisi yoktur. Tedavi semptomlara yönelik ve destek tedavisi şeklinde hastayı rahatlatmaya yöneliktir. Bu hastalık ile ilgili süren tedavi araştırmalarına aşağıdaki web sitelerinden ulaşılabilir:
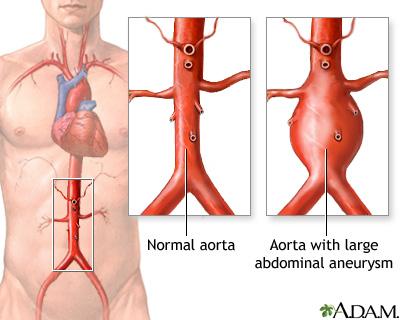
Abdominal Aort Anevrizmaları (AAA), aortun karın boşluğu kısmındaki bölümünde oluşan anevrizmalardır (damar genişlemesi). Her yaşta gerçekleşebilir ama en yaygın 50-80 yaş arası erkeklerde görülmektedir. Etkilenen çoğu birey belirti göstermezken bazılarında karın boşluğunda titreşim (nabız atması) hissi ve/veya sırt ağrısı görülebilir. Eğer anevrizma yırtılırsa çok şiddetli ağrıya, mide bulantısına, kusmaya, çarpıntıya ve şoka neden olur. AAA’ların %20’si er ya da geç yırtılır ve genelde ölümcüldür. Abdominal Aort Anevrizmalarında birden çok genetik ve çevresel faktör etkili olabilir. Bazen de kalıtsal bir sendroma bağlı olarak görülür. Bir ailede birden fazla bireyde bulunuyorsa “Ailesel Abdominal Aort Anevrizması” olarak adlandırılır.
Çoğu AAA hastasında hiç semptom görülmez ve tesadüfen başka bir amaçla yapılan karın bölgesi görüntülemesi (ultrason vb.) sırasında saptanır. Ya da fiziksel muayene sırasında abdomen stetoskopla dinlenirken anormal kan akışı sesi duyulabilir. Anevrizma genişlediği veya yırtıldığı zaman görülen en yaygın belirti, genelde karın bölgesinde başlayan ve sırta ve yanlara yayılan şiddetli ağrıdır. Diğer belirtiler anevrizmanın aortta nerede konumlandığına ve çevre yapıları etkileyip etkilemediğine bağlıdır. Yıllarca belirti göstermeden kalabilir.
Görülme Sıklığı
Abdominal aort anevrizması 60 yaş altı bireylerde nadiren görülür. 60-65 yaş arası 1000 kişiden birinde AAA görülür ve yaş arttıkça görülme sıklığı da artar. Tıbbi görüntüleme yöntemleri, AAA’ların 65 yaş üstü erkeklerin %2-13’sini; kadınlarınsa %6’sını etkilediğini göstermektedir. Ancak görüntüleme teknikleriyle belirlenen anevrizmaların neredeyse %90’ı küçük (çapı 3.5 santimden az) ve yırtılma ihtimali düşük olarak tanımlanmaktadır.
Genetik Değişiklikler
Abdominal Aort Anevrizması çok bileşenli (multifaktöriyel) bir rahatsızlıktır, yani genetik faktörler ve çevresel koşulların etkileşimiyle ortaya çıkar. Ailede AAA’ya sahip bireylerin bulunması hastalık riskini arttırır. İsveçli araştırmacılar, birinci derece yakınında AAA bulunan bir bireyde AAA gelişme ihtimalinin aile öyküsünde bu rahatsızlığın bulunmadığı bir bireye göre yaklaşık iki kat arttığını rapor etmişlerdir.
AAA kalıtsal sendromlara bağlı olarak da ortaya çıkabilir. Ehlers-Danlos Sendromu ve Marfan Sendromu, abdominal aort anevrizmasına neden olabilen, bağ doku bozukluğuyla karakterize iki hastalıktır.
Spesifik genetik değişimlerin AAA riskini arttırdığı tahmin edilse bile, sendroma bağlı olmayan AAA’ya sebep olan bir gen bilinmemektedir. Abdominal Aort Anevrizmasının kalıtımı karmaşık olduğu için bir bireyin bu rahatsızlığı geliştirip geliştirmeyeceğini öngörmek mümkün değildir.
Teşhis Yöntemleri
Fiziksel muayene AAA’yı teşhis etmek için birincil yöntemdir. Doktorun muayene sırasında karın boşluğunda pulsatil kitle hissetmesi ile teşhis konulabilir. Steteskopla dinleme sırasında da anevrizmadaki anormal kan akışı seslerinin duyulması mümkündür. Çoğunlukla ultrason vb. görüntüleme teknikleri ile AAA teşhis edilebilir ve anevrizmanın boyutu belirlenebilir.
Tedavi Yöntemleri
Abdominal Aort Anevrizması zaman geçtikçe genişler. Anevrizma büyüdükçe, yırtılması ve ölümle sonuçlanması riski de artar. Tedavi amacı anevrizma yırtılmadan önce müdahalede bulunmaktır. Semptom göstermeyen vakalarda anevrizmanın onarımının yapılıp yapılmayacağı ve ne zaman yapılacağının kararı risklere bağlı olarak değerlendirilir. Genelde çapı 4 santimden küçük olan anevrizmalarda hemen ameliyatla onarım yapılmasındansa zamanla kontrolünün yapılması önerilir. Çapı 5.5 santimden daha geniş olan veya 6 ayda 0.5 cm’den fazla genişleyen anevrizmalarda ise ameliyat önerilir.
Konjenital diseritropoetik anemi (CDA) hastalığının belirgin özelliklerinden bazıları kemik iliğinde eritroblastların etkisiz eritropoezi, hemolizi ve morfolojik anormalliklere sahip kırmızı kan hücreleridir. CDA’lar ağırlıklı olarak kemik iliğinde var olan diseritropoezin neden olduğu heterojen bir nadir konjenital anemi grubunu temsil eder.
CDA hastalığına ait sınıflandırma söz konusu olduğu zaman üç alt tipinden söz edilmektedir. Bunlar CDA tip I, CDA tip II ve CDA tip III. CDA tip I’den sorumlu olan gen CDAN1 genidir ancak bu genin fonksiyonu tam olarak anlaşılabilmiş değildir. CDA tip II’den sorumlu olan gen ise SEC23B genidir. Tip II’de sorumlu genin, son zamanlarda endoplazmik retikulum ve golgi aparatı arasındaki hücre içi keseler olan veziküller taşımaya dahil olduğu bilinen ancak eritroitteki spesifik rolü kuşkusuz ki SEC23B proteinin kodladığıdır. CDA tip III en nadir gelişen tiptir. CDA tip III ilk tanımlanan CDA tipidir fakat hastalığın genetik etiyolojisinden dolayı 60 yıl gibi bir süreden sonra değerlendirildi. KIF23 (15q21) genindeki bir mutasyondan kaynaklanmaktadır.
CDA tip I hastalarda bilim insanları KLF-1 transkripsiyonel faktörün missense mutasyonunu keşfetmişlerdir. KLF-1 eritroid transkripsiyon faktörüdür ve eritropoez için potansiyel olarak gerekli geniş bir gen spekturumunun ekspresyonunda kritik bir rol oynar. Bu yüzden insan eritroid farklılaşması sırasında KLF-1’in kilit rolünü doğrular. Ayrıca mutasyonun KLF-1 transkripsiyonel aktivitesi üzerinde baskın negatif bir etkiye sahip olduğu ve su kanalı AQP1’in ve adezyon molekülü CD44’ün ekspresyonunu beklenmedik bir şekilde ortadan kaldırdığını göstermişlerdir. KLF-1’in ekzon 3’ünde G A bazından geçişinde oluşan heterozigot missense mutasyon neden olduğu bilinmektedir. Bu mutasyon, karboksi terminalinde DNA promotör bölgeleriyle etkileşime girmek için anahtar alanlardan birisi olan çinko parmak domaininden ikincisinde KLF-1’in amino asit sekansını değiştirmektedir.
Hastalığın Yaygınlığı
CDA tip II bilinen en yaygın CDA hastalığıdır. Önceden de belirtildiği üzere CDA tip III en nadir görülen formudur. CDA tip I’in insidansı yani yaygınlığı ile ilgili bir belirsizlik veya kesinlik olmayan bir durum söz konusudur. CDA hastalığı popülasyon üzerinde çok nadir bir vakadır yeri geldiği zaman hastalığın belirtileri başka hastalıkların semptomları ile örtüştüğü için yanlış teşhis edilebiliyor.
Hastalığın Kalıtımı
CDA’nın kalıtımı kendisine bağlı alt sınıflara bağlıdır. Örneğin CDA tip I ve II otozomal resesif olarak aktarılır bu şu anlamı taşımaktadır; her hücrenin ilgili genin 2 kopyası yani allelleri dahi mutasyona uğramıştır. CDA’nın otozomal resesif formunu taşıyan bir bireyin ebeveynlerinin her biri, mutasyon geçirmiş genin bir kopyasını taşımaktadır fakat semptom veya belirti göstermeksizin yaşamlarını sürdürürler.
CDA tip III’ün kalıtım modeli olarak da otozomal dominan kalıtım modeli olarak belirtilmiştir ve bu model de mutasyona uğramış genin sadece bir tane kopyası yetecektir.
Serbest yaşayan amiplere bağlı amipiyaz tüm doğal ortamlarda otonom bir
durumda hayatta kalabilen Acanthamoeba,
Naegleria ve Balamuthia cinslerine
ait serbest yaşayan amiplerin enfeksiyonundan kaynaklanan nadir bir parazitik enfeksiyondur.
Serbest yaşayan amipler nadir ancak yıkıcı hastalıklara neden olur. Bu canlılar
dünya çapında her yerde bulunabilir. Bütün yaş gruplarından insanları etkiler
ve görülme sıklığı (prevalansı) 100.000’de 1 ila 9 kişi arasındadır.
Etken
Faktörler
Doğada pek çok cins serbest yaşayan amip bulunmaktadır, ancak bunlardan
sadece 3 tanesi insanlarda hastalık nedeni olarak tanımlanmıştır. Acanthamoeba, Naegleria ve Balamuthia.
Acanthamoeba ssp. toprakta, tozda, havada ve suda, havalandırma ve klima sistemlerinde bulunur. Acanthamoeba ve Balamuthia türleri bağışıklığı baskılanmış hastalarda kronik granülomatöz amipli ensefalite (GAE) neden olur. Ayrıca Acanthamoeba ssp., sinuzite, granülomatöz deri lezyonlarına, kontakt lens kullananlarda kornea travması sonucu amipli keratit ve korneal ülsere neden olabilir.
N.fowleri tatlı su habitatlarında bakteriler ile beslenir. Sulya ilgili
aktiviteler sırasında burun yoluyla vücuda girerek insanlarda patojeniteye
neden olabilir. Burundan girdikten sonra bu amip beyne gider ve genellikle
ölümcül bir beyin fonksiyonu olan birincil meningoensefalite (PAM) neden olur.
Granülomatöz Amipli Ensefalit:
Acanthamoeba ve Balamuthia türlerinin neden olduğu
granülomatöz amipli ensefalit (GAE) kronik ve yavaş ilerleyen bir merkezi sinir
sistemi enfeksiyondur. Bazı durumlarda hastalık akciğerleri de etkileyebilir.
Hastalığın kuluçka süresi tam olarak bilinmemekle birlikte haftalarla aylar
arası olacağı tahmin ediliyor. Genellikle aerosol yani hava yoluyla bulaşır.
Belirti ve Semptomlar
Bu hastalığın semptomları arasında, baş ağrısı, zihinsel durum
değişikliği, ateş, uyuşukluk, mide bulantısı, kusma ve bir kaç hafta içerisinde
ölüme kadar ilerleyebilen psikoz yer alabilir.
Teşhis ve Tedavi Yöntemleri
Beyin sıvısından hazırlanan kültürün PCR yöntemi ile
analiziyle teşhis edilir, ancak çok nadir görünen bir hastalık olduğu için
güvenilir bir teşhis testi bulunmamaktadır. Bu sebeple çoğu vakada ölüm
sonrasına kadar tespit edilemez. En çok immun sistemi baskılanmış hastalarda
görülür. Alkolizm, uyuşturucu kullanımı, kemoterapi, kortikosteroidler ve organ
nakli bu hastalık için risk faktörleri olarak tanımlanmıştır. Hastalığın
öldürücülük oranı neredeyse %100’dür.
Bu hastalık için yüksek iyileşme olasılığı taşıyan
kanıtlanmış bir tedavi yöntemi bulunmamaktadır. Bu sebeple tedavilerinde
yerleşik bir başarı bulunmamaktadır, ancak miltefosin, laboratuvar ortamında
birkaç serbest yaşayan amip türüne karşı öldürücü aktivite göstermekte ve son
zamanlarda miltefosin tedavisi umut verici sonuçlar vermektedir.
Kutanöz akantamoebiasis:
Acanthamoeba ve Balamuthia türlerinin neden olduğu sert nodüller ya da iyileşmeyen sertleşmiş cilt ülserleri halinde bulunan deri lezyonlarıdır. Tedavisi oldukça zordur. İtrakonazol, pentamidin ve 5-flusitozin içeren ilaçlar kullanılır. Ayrıca miltefosin tedavisi umut vaadeden sonuçlar vermektedir. Öldürücülük oranı %76 civarıdır ancak granülomatöz amipli ensefalitle birleştiğine %100’e yaklaşır.
Amipli keratit:
Acanthamoeba ssp. cinsi amiplerin sebep olduğu görme yetisini tehtit eden bir kornea hastalığıdır.Kontakt lenslerin hijyenik olmayan kullanımı, kornea aşınması ve gözün kirli suya maruz kalması sebebiyle gerçekleşebilir. Genel nüfusta 100.000 kişi içerisinde 3 kişide görülür.
Belirti ve Semptomlar
Sulanan gözler, fotofobi, göz ağrısı, bulanık görme, göz tahrişi
semptomlar arasındadır.
Teşhis ve Tedavi Yöntemleri
Kornea sıyrılması veya biyopsisi ile teşhis edilir. Tedavi için erken
teşhis önemlidir. Tedavi için ilaçların uzun süreli kullanılması gerekebilir.
Hastalıktan kaçınmak için kontakt lenserin özel sıvıları harici sularla (musluk
suyu gibi) yıkanılmasından kaçınılmalıdır.
Birincil Amipli Meningoensefalit:
N. fowleri türü amipin neden olduğu genellikle ölümcül olan, hızlı
ilerleyen bir sinir sistemi enfeksiyonudur. Kuluçka dönemi belirsizdir. Kirli
sularda bulunur. Kirli sularda yüzen kişilerde burun yoluyla merkezi sinir
sistemine geçer. İnsandan insana yayılma gösterdiğine dair herhangi bir bulgu
yoktur.
Belirti ve Semptomlar
Semptomları bakteriyel veya viral menenjitle benzerlik gösterir. Enfeksiyonlar; baş ağrısı, fotofobi, mide bulantısı ve kusma ile ilişkilendirilir. Ayrıca patojen beyin dokusunda yoğun tahribata neden olduğundan, vücutta kontrol kaybı, nöbetler ve halüsinasyonlar görülebilir. İlerleyen döneminde boyun sertliği, kraniyal sinir felci ve koma görülebilir.
Tedavisinde amfoterisin ilacı tercih edilir. Miltefosinin bu hastalık
üzerinde de kullanımı araştırılmaktadır. Hastalıktan 2011 yılına kadar kurtulan
kişi sayısı 2 olarak rapor edilmiştir. 2013 yılında miltefosinle tedavi edilen
2 çocukta da iyileşme görülmüştür. Buna rağmen hastalığın öldürücülüğü
neredeyse %100’dür
Genel Bilgi- Hastalığın Kısa Tanımı- Etken faktörler- Genetik Değişiklikler
Mevalonik asitüri, tipik olarak bebeklik döneminde başlayan tekrarlayan ateş atakları ile karakterize bir durum olan ciddi mevalonat kinaz eksikliğidir. Bu ateş olayları sırasında, mevalonik asidüri olan insanlar (büyütülmüş karaciğer ve dalak, hepatosplenomegali), lenfadenopati , karın ağrısı, ishal, eklem ağrısı ( artralji ) ve deri döküntüleri görülür. Devam eden diğer sorunlar şunlardır: gelişimsel ataksi, görme ile ilgili problemler, alışılmadık derecede küçük ve uzun bir kafa ve gelişememe sorunu. Mevalonik asidüri, mevalonat kinaz eksikliğinden kaynaklanır. Bu eksiklik, kalıtımsal mutasyonlar sebebiyle MVK geninden kaynaklanır. Tedavi zordur ve esas olarak destekleyicidir. Daha az şiddetli mevalonat kinaz eksikliğine hiperimmunoglobulinemi D sendromu (HIDS) denir.
Belirti ve Semptomlar
Çoğu hastalık için semptomlar kişiden kişiye değişir. Aynı hastalığı olan insanlar listelenen tüm semptomlara sahip olmayabilir. Mevalonik asidüri , hepatosplenomegali, lenfadenopati, artralji ve deri döküntüsü eşliğinde dismorfoloji, psikomotor gerilik, ilerleyici serebellar ataksi ve tekrarlayan ateşli krizlerle karakterizedir Bu bilgiler İnsan Fenotip Ontolojisi (HPO) adı verilen bir veritabanından alınmıştır
Tıbbi terimler
Diğer isimler
Serebral kortikal at-rofi
Beyin hücrelerinin kaybı nedeniyle beynin dış tabakasının boyutunda azalma
Gecikmiş iskelet ol- gunlaşması
Gecikmiş kemik olgunlaşması
dolikosefali
Uzun, dar kafa
Aşağı eğimli palpeb-ral çatlaklar
Göz kapakları arasındaki açıklığın aşağı doğru eğilmesi
Global gelişimsel ge-cikme
Genetik Görülme Sıklığı
Kesin bir bilgi bulunmamaktadır.
Kalıtım Paterni/Deseni
Yer
Fenotip
Fenotip MIM nu-marası
Kalıtım
Fenotip Eşleme Anahta-rı
Gen / Lokus
Gen / Lokus MIM numa– rası
12q24.11
Mevalonik asitüri
610377
AR
3
MVK
251170
Mevalonik asidüri (MEVA), 12q24 kromozomundaki mevalonat kinaz genindeki (MVK; 251170) homozigot veya bileşik heterozigot mutasyondan kaynaklanır. Otozomal resesif gen kaynaklıdır.
Teşhis Yöntemi ve Tedaviler
Genetik veya nadir bir hastalık için teşhis koymak genellikle zor olabilir. Sağlık mesleği mensupları tanı koymak için genellikle bir kişinin tıbbi geçmişine, semptomlarına, fizik muayenesine ve laboratuvar test sonuçlarına bakar. Aşağıdaki kaynaklar, bu durumun teşhisi ve testi ile ilgili bilgi sağlar. Teşhis hakkında sorularınız varsa, bir sağlık uzmanıyla görüşmelisiniz.
Test Kaynakları
Genetik Test Kayıt (GTR) bu durum için genetik testler konusunda bilgi sağlar. GTR için hedef kitle sağlık hizmeti sağlayıcıları ve araştırmacılardır. Genetik test hakkında özel soruları olan hastalar ve tüketiciler bir sağlık uzmanına veya bir genetik uzmanına başvurmalıdır.
Tüm hastalarda etkili olan standart bir tedavi yoktur, bu nedenle esas olarak destekleyici bir reçete uygulanır. Simvastatin, mevalonik asitüri olan iki hastanın klinik durumunu kötüleştirmiştir. HIDS hastalarında bir dereceye kadar başarı ile kullanılan başka bir ilaç olan Anakinra, kısmi olarak indüklemiştir. Ancak tüm hastalar bu kadar olumlu yanıt vermez.
Allojenik kemik iliği transplantasyonu ile mevalonik asidürinin başarılı bir şekilde tedavi edildiğine dair raporlar da ortaya çıkmıştır. Bu noktada, bu tedavi hala araştırılıyor ve potansiyel olarak anti-enflamatuar ilaçlarla (örn. TNF-alfa ve interlökin-1 beta inhibitörleri) tedaviye dirençli mevalonik asidüri hastalarına uygulanabilir.
Hastalıkla İlişkili Genler
Kalıtımsal mutasyonlar sebebiyle MVK geninden kaynaklanır.
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Kromoblastomikoz, deri ve deri altı dokuyu etkileyen kabarık
ve kabuklu lezyonlarla ilişkilendirilimiş kronik bir mantar (genellikle Fonsecaea
Pedrosoi3) enfeksiyonudur. Çoğunlukla kol,bacak gibi uzuvlarda
görülür, ancak vücudun herhangi bir bölgesini etkileyebilir. Kromoblastomikoz,
toprakta, tahtada ve/veya çürüyen bitki içerisinde bulunan birkaç mantardan
kaynaklanmaktadır. Genellikle kıymık gibi küçük bir yaralanma yoluyla cilde
girer1. Tropikal ve subtropikal iklime sahip en yaygın alanlardır2.
Kromoblastomikoz tedavisi, itrakonazol ve flusitozin gibi ilaçları, kriyoterapi
veya ameliyatı içerebilir1,2.
Belirti ve Semptomlar
Klinik
Bulgulara bakıldığında hastalığın ilerlemesi sürecinde gerçekleşen belirti ve
semptomlar sırasıyla aşağıda verilmiştir ;
Küçük, sert kırmızı veya gri bir yumru olarak başlar1.
Yılda sadece 2 mm gibi çok küçük bir büyüme oranı göstermektedir1.
Sonunda, siğilli kuru bir nodül veya plak gelişir1.
Lezyonun merkezinde en azından kısmi yara izi oluşabilir1.
Etkilenen uzuv genellikle büyüyebilir (fil hastalığı)1.
İlkinin etrafında zamanla yeni lezyonlar gelişebilir veya enfeksiyon yeni bir bölgeye taşınabilir1.
Hiçbir rahatsızlığa neden olmayabilir ancak genellikle aşırı kaşıntı gözlemlenmektedir1.
Nadiren, uzun süredir devam eden kromoblastomikoz nedeniyle Skuamöz Hücreli Karsinom (SCC) oluşumu gerçekleşir1.
Hastalığın genetic olarak değerlendirilmesini ele alan bir
çalışma yapılmamış,dolayısıyla bir bilgi bulunmamaktadır.
Kalıtım Paterni/Deseni
Hastalığın kalıtım
paterni hakkında bir çalışma yapılmamış,dolayısıyla bir bilgi bulunmamaktadır.
Teşhis Yöntemleri ve Tedaviler
Teşhis
Kromoblastomikoz
histopatolojisi, cilt biyopsisinde, dematiase bir mantarın varlığını doğrulayan
tipik kalın duvarlı koyu kahverengi “sklerotik” hücreler
gösterebilir. Organizmanın duvarlarında bulunan melanin nedeniyle koyu renklidir1.
Sabouraud besiyerinde 25-30 C° antibiyotikli kültür, bir veya iki hafta sonra zeytin yeşili ila siyah renkte mantar kolonileri büyür. Sorumlu mantarı isimlendirmek zor olduğundan dematiaceous mantarların neden olduğu enfeksiyon Phaeohyphomycosis olarak isimlendirilerek kullanılmaktadır1.
2.Tedavi
Nadiren,
kromoblastomikoz kendiliğinden iyileşerek bir yara izi bırakır.
Tedavisi zor ve
uzun olmakla beraber şunları içerebilir1:
İtrakonazol,
posakonazol veya vorikonazol, muhtemelen terbinafin ile kombinasyon halinde,
Flusitozin,
Tiyabendazol,
Local
Isıtma,
Kriyoterapi,
Etkilenen
dokuyu tamamen çıkarmak için yapılan ameliyat1.
Hastalıkla İlişkili Genler
Bilinmiyor.
Hastalığın Diğer İsimleri
Kromomikoz – Kromoblastomikoz
Hastalıkla Benzer
Özellik Gösteren(Karıştırılan) Diğer Hastalıklar
Sporotrikoz gibi diğer mantar
enfeksiyonları,
Atipik mikobakteri enfeksiyonu,
tüberküloz, cüzzam ve sifiliz gibi bakteriyel enfeksiyonlar,
Leishmaniasis gibi protozoal enfeksiyonlar,
Skuamöz hücre karsinoması,
Sedef hastalığı, diskoid lupus
eritematoz gibi cilt bozuklukları1.
Arguello-Guerra, L., Gatica-Torres, M., &
Dominguez-Cherit, J. (2016). Chromomycosis. BMJ case reports, 2016,
bcr2016215391. https://doi.org/10.1136/bcr-2016-215391