Havva Özserçe
Genel Bilgi
Börjeson-Forssman-Lehmann sendromu, zihinsel engellilik, endokrin bozukluklarına bağlı obezite ve büyüme kusurlarına neden olan nadir bir genetik hastalıktır.
Börjeson-Forssman-Lehmann sendromu (BFLS), zihinsel sakatlık, obezite, nöbetler, erkeklerde testislerin veya kadınlarda yumurtalıkların hormon üretmemesi (hipogonadizm) ve ayırt edici yüz özellikleri ile karakterize oldukça nadir görülen bir hastalıktır. Etkilenen bebekler genellikle gelişimsel dönüm noktalarına ulaşmada gecikmeler yaşarlar. Kesin semptomlar, aynı ailenin üyeleri arasında bile durumdan duruma değişir.
BFLS’den etkilenen erkeklerin çoğu, değişen şiddette zihinsel gerilik ile karakterizedir. Bazı durumlarda nöbetler olabilir. Hafif obezite etkilenen çocuklarda bebeklik döneminde bile yaygındır.
BFLS, X kromozomu üzerinde PHF6 geninin bozulmasından veya değişikliklerinden (mutasyonlar) kaynaklanır.
Genetik Değişiklikler /Etken Faktörler
Börjeson-Forssman-Lehmann sendromu, erkeklerde ve kadınlarda tam olarak ifade edilebilen nadir bir hastalıktır. Kadınlarda fenotipik ifade oldukça değişkendir ve henüz tahmin edilemez. BFLS başlangıçta tıbbi literatürde 1962’de üç ilgili erkekte ve daha az etkilenen kadın akrabalarından üçünde tanımlanmıştır. BFLS’nin kesin insidansı bilinmemektedir, ancak tek tek etkilenen kadınların tanımlanmasıyla, belirsiz olduğunu varsayıyoruz. PHF6 geninde mutasyonlar bulunan yaklaşık 40 alakasız aile ve çeşitli izole vakalar (erkekler ve artan sayıda kadın) bildirilmiştir. Bununla birlikte, PHF6 mutasyonuna sahip gerçek birey sayısının, teşhis edilen vakaların bile rapor edilmediği ve dolayısıyla yakalanmadığı kadar yüksek olduğu düşünülebilir .
Belirti ve Semptomlar
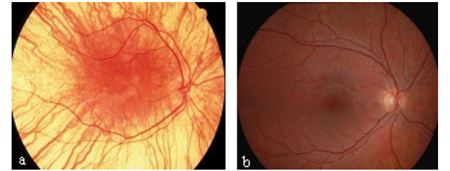
Etkilenen bireyler, büyük, etli kulak memeleri, derin ayarlı gözler, gözlerin üstündeki ağır sırtlar (göze çarpan supraorbital sırt) ve yüzün kalınlaşmış bağ dokusu gibi yüze kaba bir görünüm veren ayırt edici yüz özelliklerine sahip olabilir. Bazı durumlarda, etkilenen bireylerde üst göz kapaklarında sarkma (pitoz), hızlı, istemsiz göz hareketleri (nistagmus) ve gözlerin arkasını (retinanın) hatlayan ince zarın ve ana sinirleri beyine retina (optik sinir). Uzak görüşlülük (hipermetrop) ve katarakt gibi görme sorunları 30 yaşından önce gelişebilir.
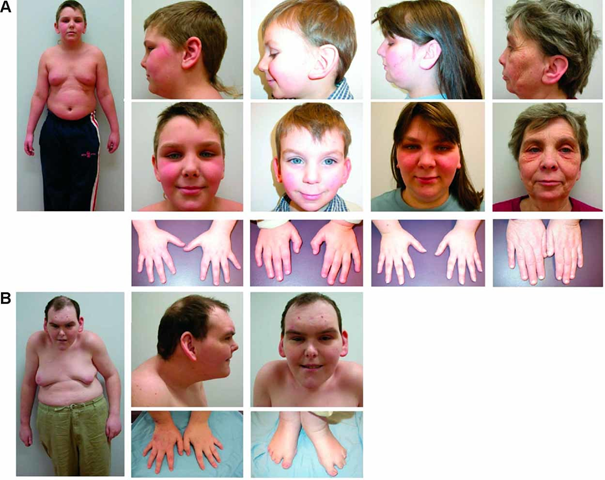
BFLS’li bireylerde testis veya yumurtalıkların (hipogonadizm) işlevi de azalmış olabilir. Testislerin ve yumurtalıkların hormon üretmemesi, kısa boy ve gecikmiş cinsel gelişim ile sonuçlanan büyüme eksikliklerine neden olabilir. Ek olarak, etkilenen erkeklerde küçük genital bölgeler olabilir ve testisler skrotuma (kriptorşidizm) inmeye başarısız olabilir. Ergenlikten sonra, bazı erkeklerde anormal şekilde büyümüş göğüsler (jinekomasti) gelişebilir.
İskelet anormallikleri, omurganın anormal yan yana veya önden arkaya eğriliği (skolyoz veya kifoz), dar bir servikal spinal kanal veya ortaya çıkan bazı parmak veya ayak parmaklarının az gelişmişliği (hipoplazi) gibi bazı durumlarda ortaya çıkabilir. uzun, konik parmaklar ve anormal derecede kısa ayak parmakları, özellikle dördüncü ve beşinci ayak parmakları.
Etkilenen erkekler yaşlandıkça, bozukluğun semptomları daha hafif hale gelebilir ve vakalar arasında daha fazla değişebilir. BFLS’li bazı erişkinlerde diyabet meydana gelmiştir.
BFLS için hastalık genini taşıyan kadınların çok daha az ciddi bir şekilde etkilendiği ve sadece etkilenen erkeklerde görülen daha hafif bir BFLS formu olan bozukluğun sadece bazı semptomlarını geliştirdiği düşünülmüştür. Bununla birlikte, bu varsayım, esas olarak etkilenen erkekleri olan daha büyük ailelerden gelen kadınlara dayanıyordu. Yeni DNA sekanslama teknolojilerinin ortaya çıkmasıyla birçok singleton hastası test edildi ve son zamanlarda PHF6’daki de novo mutasyonlarının (çocuğun ebeveynlerinde mevcut olmayan) tekli kadınlarda spesifik bir klinik fenotipe neden olduğu ortaya çıktı. şimdiye kadar az tanınmıştı. Bu kadın hastalar değişken düzeyde zihinsel engellilik, karakteristik yüz özellikleri, az gelişmiş tırnaklar, bazı diş anomalileri, seyrek saçlar ve doğrusal cilt hiperpigmentasyonundan etkilenir. Birkaç kadının, MRG’de subkortikal bant heterotopisine benzeyen genel bir nöronal migrasyon bozukluğu belirtileri ile ilişkili olabilen epilepsi geliştirdiği bildirilmiştir. İlginç bir şekilde, bu kadın klinik fenotipi BFLS ile örtüşürken, ek klinik özellikler de içerir, böylece hastalığa yeni bir yön ekler. Etkilenen bu dişiler (özellikle gençken) başka bir genetik sendrom olan Coffin-Siris sendromuna benziyor gibi görünüyor.
Genetik Görülme Sıklığı
Börjeson-Forssman-Lehmann sendromunun yaygınlığı bilinmemektedir, oldukça nadir görülen bir bozukluk olup, 24 aileden ve sporadik vakadan yaklaşık 50 hasta bildirilmiştir.
Kalıtım Paterni / Deseni
BFLS, hücre büyümesi ve proliferasyonunda yer alan bir protein olan PHF6’yı kodlayan Xq26 üzerinde bulunan PHF6 genindeki mutasyonlardan kaynaklanır . Genel olarak, PHF6 ekspresyonunun kaybı veya azalması ile sonuçlanan mutasyonlar daha ciddi klinik semptomlara neden olur.
Teşkis Yöntemleri ve Tedavileri
Teşhis
Börjeson-Forssman-Lehmann sendromu tanısı, kapsamlı bir klinik değerlendirme, ayrıntılı bir hasta öyküsü ve karakteristik özelliklerin tanımlanması temelinde konur. İskeletin röntgeni (iskelet radyografisi), potansiyel iskelet defektlerinin varlığını tespit etmek ve şiddetini değerlendirmek ve BFLS tanısını desteklemek için kullanılabilir. Tanıyı doğrulamak için PHF6 genindeki mutasyonlar için moleküler genetik test mevcuttur.
Bir ebeveynde bir PHF6 mutasyonunun olduğu biliniyorsa, doğum öncesi tanı mümkündür.
Tedavi
BFLS için bir tedavi yoktur, tedavisi, her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, bir uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, nörologlar, göz uzmanları (oftalmologlar) ve iskelet bozuklukları (ortopedistler) ve diğer sağlık uzmanlarının tedavisinde etkilenen bir çocuğun tedavisini sistematik ve kapsamlı bir şekilde planlaması gerekebilir.
Erken gelişimsel müdahale, BFLS’li etkilenen çocukların potansiyellerine ulaşmalarını sağlamada önemlidir. Etkilenen çocuklara faydalı olabilecek özel hizmetler, özel iyileştirici eğitim ve diğer tıbbi, sosyal ve / veya mesleki hizmetleri içerebilir. Genetik danışmanlık etkilenen bireyler ve aileleri için yararlı olabilir.
BFLS yaşamı tehdit edici değildir, ancak karşılaşılan zihinsel engellilik nedeniyle yaşam kalitesi sınırlıdır ve yaşam boyu denetim genellikle gereklidir.
Hastalığın Diğer İsimleri
- BFLS
- BORJ
- Borjeson sendromu
Kaynak
- https://rarediseases.org/rare-diseases/borjeson-forssman-lehman-syndrome/
- https://www.omim.org/entry/301900
- https://rarediseases.info.nih.gov/diseases/936/borjeson-forssman-lehmann-syndrome
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=127
- https://accessanesthesiology.mhmedical.com/content.aspx?bookid=852§ionid=49517303











{kind=link}
{kind=link}
{kind=link}
{kind=link}