Aspartilglukosaminüri , merkezi sinir
sistemini kapsayan ve iskelet anormalliklerine ve bağ dokusu lezyonlarına neden
olan ciddi otozomal resesif lizozomal depo bozukluğudur. En karakteristik
özelliği ilerleyici zihinsel geriliktir. Bozukluğa, lizozomal enzim
glikosilasparaginazın yetersiz aktivitesi neden olur; AGU, genellikle
Finlandiya hastalık mirası olarak adlandırılan bozukluklar grubuna aittir.
Belirtiler
Aspartilglikozaminüri olan bebekler doğumda
sağlıklı görünür ve gelişim çocukluk boyunca tipik olarak normaldir. 2 veya 3
yaşlarında ortaya çıkan bu durumun ilk işareti genellikle konuşma gecikmesidir.
Hafif zihinsel engelli belirtisi daha sonra belirginleşir ve öğrenme yavaş bir
hızda gerçekleşir. Entelektüel sakatlık ergenlik döneminde giderek kötüleşir.
Bu bozukluğu olan çoğu insan öğrendikleri konuşma yeteneğinin çoğunu kaybeder
ve etkilenen yetişkinlerin kelime dağarcığında genellikle sadece birkaç kelime
vardır. Aspartilglukozaminüri olan yetişkinlerde nöbet veya hareketle ilgili
sorunlar gelişebilir. Bu durumdaki insanlarda giderek zayıflayan ve kırılmaya
müsait hale gelen kemikler
(olabilirosteoporoz), alışılmadık derecede geniş bir eklem hareketi (
hipermobilite ) ve gevşek cilt semptomları görülür. Etkilenen bireyler, geniş
aralıklı gözler ( oküler hipertelorizm ), küçük kulaklar ve dolgun dudaklar
içeren karakteristik bir yüz görünümüne sahiptir . Burun kısa ve geniştir ve
yüz genellikle kare şeklindedir. Bu duruma sahip çocuklar yaşları için uzun
olabilir, ancak ergenlik döneminde büyüme hamlesi olmaması tipik olarak
yetişkinlerin kısa olmasına neden olur. Etkilenen çocuklar da sık sık üst
solunum yolu enfeksiyonu geçirme eğilimindedir. Aspartilglukozaminüri olan
bireyler genellikle yetişkinliğin ortalarına kadar hayatta kalırlar. İskelet
deforme olabilir. Omurga bükülmüş olabilir (skolyoz) ve boyun alışılmadık
derecede kısa olabilir. Gözler de katarakt gelişebilir. Davranış sorunları
yaygındır. Akciğer, kalp ve kan problemleri sonraki yıllarda ortaya çıkma
eğilimindedir. Bireylerde %80-%90 görülen belirtiler;
amino asit metabolizmasında anormallik
idrarda yüksek aspartilglukozamin seviyeleri
Gecikmiş konuşma ve dil gelişimi
İstemsiz kas hareketleri bozukluğu
Dişeti büyümesi
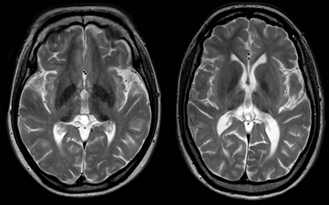
[1.1] İki aspartilglikozaminüri hastası 3.0 T olarak görüntülendi. Solda 34 yaşında kadın hasta, sağda 26 yaşında kadın hasta. Pulvinar çekirdeklerde tipik sinyal yoğunluğu azalması gösteren eksenel T2 ağırlıklı görüntüler. Her ikisinde de, gri ve beyaz madde arasındaki zayıf farklılaşma, sinyal yoğunluğu artışı olan bazı düzensiz joktakortikal odaklar, 3. ventrikülün hafif dilatasyonu, bir epifiz kisti ve nispeten kalın bir kafatası da görülebilir.
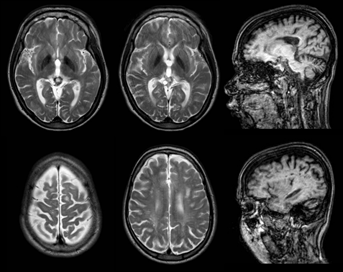
[1.2] 33 yaşında kadın aspartilglikozaminüri hastası, 3.0 T’de görüntülendi. Solda, daha önce aspartilglikozaminüri ‘da bildirilmeyen talamide atipik sinyal yoğunluğu artışı gösteren dört eksenel T2 ağırlıklı görüntü ve periventriküler ve juktakortikal beyaz maddede sinyal yoğunluğu artışı olan birkaç düzensiz odak var. Sağda pulvinar çekirdeğindeki SI azalmasını ve beyaz maddedeki bazı fokal hipertansiyon lezyonlarını gösteren iki sagital T1 ağırlıklı görüntü vardır. Ayrıca bu hastanın görüntülerinde hafif lateral ve 3. ventrikül dilatasyonu, gri ve beyaz madde arasındaki zayıf farklılaşma, hafif dilate perivasküler boşluklar, pineal kist ve hafif genel atrofi görülebilir.
Nedenler
AGA genindeki mutasyonlar aspartilglukosaminüriye
neden olur. AGA geni, aspartylglucosaminidase olarak adlandırılan bir enzim
üretmek için talimatlar içerir. Bu enzim, geri dönüşüm merkezleri olarak işlev
gören hücrelerdeki lizozomlarda aktiftir. Lizozomlar içindeki enzim, belirli
proteinlere (glikoproteinler) bağlı şeker molekülleri (oligosakkaritler)
komplekslerinin parçalanmasına yardımcı olur. AGA gen mutasyonları,
lizozomlarda aspartilglukosaminidaz enziminin yokluğuna veya eksikliğine neden
olarak glikoproteinlerin normal parçalanmasını önler. Sonuç olarak, lizozomlar
içinde glikoproteinler birikebilir. Fazla glikoproteinler hücrenin normal
fonksiyonlarını bozar ve hücrenin tahrip olmasına neden olabilir. Bir
glikoprotein birikimi özellikle beyindeki sinir hücrelerini etkilemektedir; bu
hücrelerin kaybı aspartilglukosaminüri belirtilerinin ve semptomlarının çoğuna
neden olur .
Epidemiyolojisi
Aspartilglikozaminüri ‘nın Finlandiya’daki 18.500 kişiden 1’ini etkilediği tahmin edilmektedir. Bu durum Finlandiya dışında daha az yaygındır, ancak oranı bilinmemektedir.
Kalıtım Kalıbı
Bu durum otozomal resesif paternde kalıtsaldır
yani her hücredeki genin her iki kopyasının mutasyonları vardır. Otozomal
resesif koşulu olan bir bireyin ebeveynlerinin her biri mutasyona uğramış genin
bir kopyasını taşır, ancak tipik olarak durumun belirtilerini ve semptomlarını
göstermezler.
Teşhis Yöntemleri
Biyokimyasal olarak, bir amino asit veya oligosakkarit
kromatografisi üzerine aspartilglikozaminin çokça bulunduğu idrarın atılımı ile
karakterizedir. Sonuçlar, lenfositler, fibroblastlar, amniyositler veya
trofoblastta ölçülen düşük aspartilglikozaminidaz aktivitesi ile doğrulanır.
Tedavi
Bu güne kadar tedavi edici tek girişim, 5 Fin
hastası ile sınırlı allojenik kemik iliği aşılama işlemiydi.
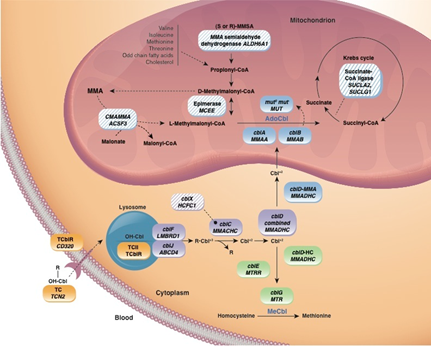
B12
bağımlı bir enzim olan metilmalonil-KoA mutaz enziminin eksikliğinin yol açtığı
otozomal resesif geçişli bir metabolik hastalıktır. Bu enzim
Metilmalonil-KoA’nın süksinilKoA’ya dönüşümünü sağlaryani bu enzim yoksa
metilmalonil-KoA süksinilKoA. ya dönüşemeyeceğinden dolayı birikir kanda
düzeyi çok artınca da idrarda metilmalonik asit atılımı gerçekleşir.
Hastalık kalıtsal olup otozamal resesif genler ile taşınmaktadır.
Belirti ve Semptomlar
Belirtiler yeni doğan
döneminden erişkin dönemine kadar devam eder. Yaşamın çok erken döneminde 1 ila
4 hafta arasında bu belirtiler anında kendisini gösterir. Genel olarak hastalık kendiliğini yaşamın ilk
anlarından itibaren kusma , gelişmede gerilik , kas hipotoni ,katoasidosizle kendisini belli eder. Hastalar anemi belirtisi de
gösterebilir. Sıklıkla akut ve ölümcül
olup bazı bireylerde kronik olarak devam eder.
Görülme Sıklığı
Türkiye’de sıklığı tam
olarak bilinmemektedir. Akraba evliliği olan hastalarda olma ihtimali
yüksektir. Bugüne kadar 450 den fazla vaka teşhis edilmiştir.
Hastalıkla İlgili Genler
MMUT , MMAA , MMAB , MCEE ve MMADHC bu
beş genin bir tanesinde anne ve babada taşıyıcı olması durumunda tanı
konulabilir.
Tedavi / Teşhis
Kritik hasta bireyler, hacim durumu ve asit-baz dengesini geri yükleyerek stabilize edilir; protein alımının azaltılması veya ortadan kaldırılması; katabolizmayı durdurmak için yüksek glikoz içeren sıvılar ve insülin yoluyla artan kalorilerin sağlanması; ve serum elektrolitleri ve amonyak, venöz veya arteriyel kan gazları ve idrar çıkışının izlenmesi. Yönetim, propiojenik amino asit öncülerinde düşük kalorili bir diyet içerir; hidroksokobalamin kas içi enjeksiyonları; karnitin takviyesi; bağırsak florasından propionat üretimini azaltmak için neomisin veya metronidazol gibi antibiyotikler; gerektiği gibi gastrostomi tüpünün yerleştirilmesi; ve enfeksiyonların agresif tedavisi. Sınırlı sayıda hastada kullanılan diğer terapiler arasında akut hiperammonemik atakların tedavisi için N- karbamilgluktamat; karaciğer, böbrek veya kombine karaciğer ve böbrek nakli; ve optik sinir atrofisinin tedavisi için antioksidanlar gibi yöntemler kullanılabilir.
Hastalıkta Kaçınılması Gereken
Oruç ve artan diyet proteinlerinden uzak durulması tavsiye edilir.
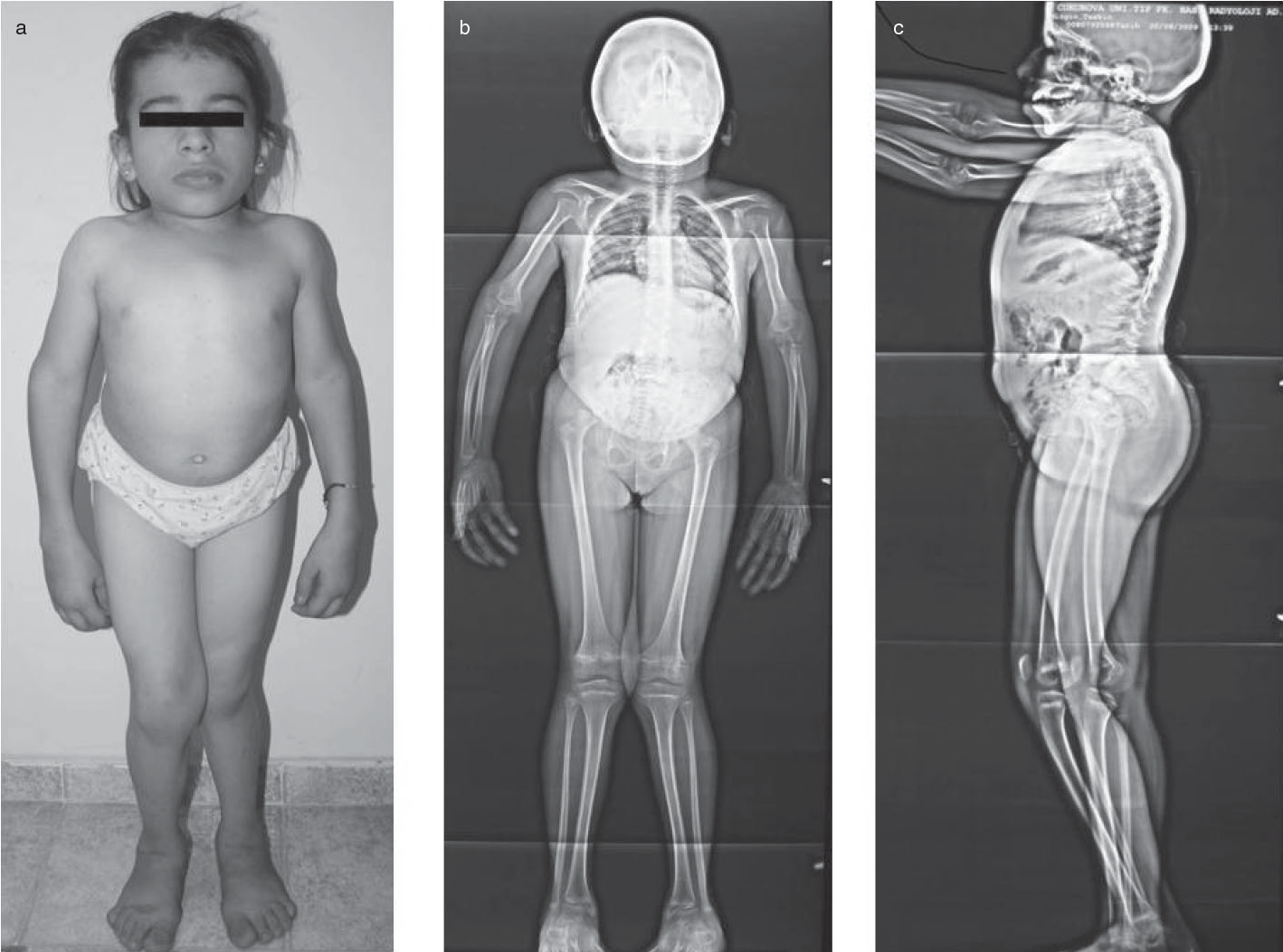
Turner sendromu, kızlar arasında en çok görülen kromozomal anomalilerden biridir. Nedeni, dişilerde 2 adet olması gereken X kromozomlarından birinin yokluğu ya da anormal olmasıdır. En tipik belirtileri, geniş ya da yele (perdeli) boyun (ekstra deri kıvrımları), bebeklerde şişmiş el ve ayaklar (lenfödem) , düz ve dışa dönük tırnaklar, iskelet anormallikleri veya böbrek problemleri vardır. Turner sendromlu bireylerin üçte biri ila yarısı, kalpten çıkan büyük arterin daralması (aort koarktasyonu) veya aortu kalbe (aort kapak) bağlayan kapak anormallikleri gibi bir kalp kusuru ile doğar. Bu kalp kusurları ile ilişkili komplikasyonlar hayatı tehdit edici olabilir. Turner sendromunun en yaygın özelliği, yaklaşık 5 yaşlarında ortaya çıkan kısa boydur. Erken yumurtalık fonksiyon kaybı (yumurtalık hipofonksiyonu veya erken yumurtalık yetmezliği) de çok yaygındır. Yumurtalıklar ilk başta normal olarak gelişir, ancak yumurta hücreleri (oositler) genellikle erken ölür ve çoğu yumurtalık dokusu doğumdan önce dejenere olur. Turner sendromlu çoğu kız ve kadın normal zekaya sahiptir. Bu özellikler etkilenen bireyler arasında farklılık gösterse de, gelişimsel gecikmeler, sözsüz öğrenme güçlükleri ve davranış problemleri mümkündür.
TS ilk olarak 1938’de Amerika Birleşik
Devletleri’nde Dr. Henry Turner tarafından tanımlanmıştır.
Genetik Değişiklikler /Etken Faktörler
Turner sendromu, iki cinsiyet
kromozomundan biri olan X kromozomu ile ilişkilidir. İnsanlar tipik olarak her
hücrede iki cinsiyet kromozomuna sahiptir: dişilerde iki X kromozomu
bulunurken, erkeklerde bir X kromozomu ve bir Y kromozomu bulunur. Turner
Sendromu, bir dişi hücrelerinde normal bir X kromozomu bulunduğunda ve diğer
cinsiyet kromozomu eksik olduğunda veya yapısal olarak değiştiğinde ortaya
çıkar. Eksik genetik materyal doğumdan önce ve sonra gelişimi etkiler.
Turner sendromlu bireylerin yaklaşık
yarısında monozomi X vardır, bu da bireyin vücudundaki her hücrenin normal iki
cinsiyet kromozomu yerine X kromozomunun yalnızca bir kopyasına sahip olduğu
anlamına gelir.
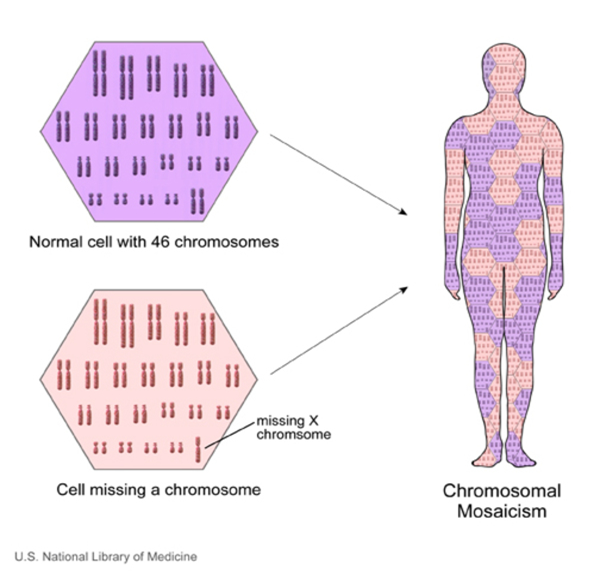
Turner sendromlu bazı kadınlar,
hücrelerinin sadece bazılarında mozaikçilik olarak bilinen kromozomal bir
değişikliğe sahiptir. X kromozomu mozaikçiliğinin neden olduğu Turner sendromlu
kadınların mozaik Turner sendromuna sahip olduğu söylenir.
Turner sendromunun
semptomları ve şiddeti bir kişiden diğerine oldukça değişebilir. Bozukluğun
birçok özelliği spesifik değildir ve diğerleri zamanla yavaşça gelişebilir veya
daha az belirgin olabilir. Etkilenen bireylerin aşağıda tartışılan semptomların
hepsine sahip olmayabileceğini belirtmek önemlidir. Etkilenen bireyler, özel
vakaları, ilişkili semptomları ve genel prognozu hakkında doktorları ve tıbbi
ekipleriyle konuşmalıdır.
Turner sendromlu hemen hemen
tüm kadınlar büyüme başarısızlığı gösterir ve ortalamadan daha kısa bir son
boyuta (kısa boy) ulaşır. Çocuklar başlangıçta, genellikle yaşamın ilk birkaç
yılında normal büyüme gösterebilir. Bununla birlikte, çoğu durumda, büyüme
oranı nihayetinde normalden daha yavaş olur ve etkilenen çocuklar normal büyüme
atakları yaşamazlar (örneğin ergenlik döneminde büyüme fışkırması olmaz).
Turner sendromunun bir diğer
yaygın özelliği, yumurtalıkların düzgün gelişememesidir (gonadal disgenez).
Gonadal disgenez, çocukluk döneminde erken yumurtalık fonksiyonunun kaybına
neden olabilir (erken yumurtalık yetmezliği). Normal olarak, yumurtalıklar
ergenlikte seks hormonları (örn. Östrojen ve progesteron) üretir. Bu hormonlar
ergenliğin başlaması ve ikincil cinsel özelliklerin doğru gelişimi için
gereklidir. Etkilenen kadınların çoğu, göğüsleri ve normal kadın vücudu
hatlarını geliştirmek, uygun kemik büyümesine girmek ve adet görmeye başlamak
için hormon replasman tedavisine ihtiyaç duyacaktır. Bazı durumlarda, etkilenen
bireyler meme gelişimine girebilir ve terapi olmadan menstruasyona başlayabilir
(spontan pubertal gelişim), ancak çoğu cinsel gelişimini durduracak ve gençlik
yıllarında daha sonraki bir noktada adet görmeyi durduracaktır.
Turner sendromlu dişiler,
perdeli bir görünüme sahip kısa bir boyun, başın arkasındaki düşük bir saç çizgisi,
düşük ayarlı kulaklar ve yukarı doğru çevrilmiş dar tırnaklar ve ayak
tırnakları gibi çeşitli farklı fiziksel özellikler geliştirebilir. Bazen
“kalkan sandığı” olarak adlandırılan geniş aralıklı meme uçlarına sahip geniş
bir göğüs oluşabilir. Bazı bireylerin şişmiş, kabarık elleri ve ayakları
olabilir. Bu belirtiler, lenfatik sistemi etkileyen bir durum olan lenfödem
nedeniyle ortaya çıkabilir. Lenfatik sistem, vücutta belirli protein açısından
zengin sıvı (lenf) ve kan hücrelerini filtreleyen ve dağıtan damarlar, kanallar
ve düğümlerin dolaşım ağıdır. Lenfödem, vücudun etkilenen bölgelerindeki sıvı
birikimine (ödem) bağlı şişme ile karakterizedir.
Ek fiziksel bulgular
arasında bir çene (retrognati), çapraz gözler (şaşılık), tembel gözler
(ambliyopi), sarkık göz kapakları ve ağızda darlık, yüksek kemerli bir çatı
(damak) sayılabilir. Bazı bireylerde ellerin kısa kemikleri, özellikle dördüncü
metakarplar, dirseklerde ortaya çıkan kollar ve düz ayaklar (pes planus) dahil
iskelet malformasyonları olabilir. Olguların yaklaşık% 10’unda, omurganın
anormal yanal eğriliği (skolyoz) de ortaya çıkabilir.
Bazı Turner sendromu
vakalarıyla ilişkili kalp kusurları, akciğerlerin arterlerinin yüksek tansiyonu
(pulmoner hipertansiyon) veya içte bir yırtık olan aort diseksiyonu da dahil
olmak üzere ciddi, hayatı tehdit eden komplikasyon riskini artırabilir.
Genetik Görülme Sıklığı
Bu durum dünya çapında 2.500 yeni doğan
kızdan yaklaşık 1’inde görülür, ancak terimde hayatta kalmayan hamileliklerde
(düşükler ve ölü doğumlar) çok daha yaygındır. Bozukluğun sıklığını etkileyen
bilinen herhangi bir ırksal veya etnik faktör yoktur. Bazı durumlarda, bozukluk
doğumdan önce veya doğumdan kısa bir süre sonra teşhis edilir. Bununla
birlikte, hafif vakalar yaşamın ilerleyen dönemlerine kadar ve hatta
yetişkinlik döneminde bile teşhis edilmeden kalabilir.
Kalıtım Paterni / Deseni
Turner sendromu vakalarının
çoğu kalıtsal değildir. Bu durum monozomi X’ten kaynaklandığında, kromozomal
anormallik, etkilenen kişinin ebeveyninde üreme hücrelerinin (yumurta ve sperm)
oluşumu sırasında rastgele bir olay olarak ortaya çıkar. Ayrılma adı verilen
hücre bölünmesindeki bir hata, anormal sayıda kromozomlu üreme hücrelerine
neden olabilir. Örneğin, bir yumurta veya sperm hücresi, ayrılmanın bir sonucu
olarak bir cinsiyet kromozomunu kaybedebilir. Bu atipik üreme hücrelerinden
biri çocuğun genetik yapısına katkıda bulunursa, çocuk her hücrede tek bir X
kromozomuna sahip olacak ve diğer cinsiyet kromozomunu kaçırmayacaktır.
Mozaik Turner sendromu da
kalıtsal değildir. Etkilenen bir kişide, erken fetal gelişimde hücre bölünmesi
sırasında rastgele bir olay olarak ortaya çıkar. Sonuç olarak, etkilenen bir
kişinin hücrelerinin bazıları normal iki cinsiyet kromozomuna sahiptir ve diğer
hücrelerde X kromozomunun yalnızca bir kopyası bulunur. X kromozom mozaikliği
olan kadınlarda diğer cinsiyet kromozom anormallikleri de mümkündür.
Nadiren, X kromozomunun
kısmen silinmesinin neden olduğu Turner sendromu bir nesilden diğerine
geçebilir.
Aşağıdaki bozuklukların
belirtileri Turner sendromunun semptomlarına benzer olabilir. Karşılaştırmalar
ayırıcı tanı için yararlı olabilir.
Noonan sendromu, doğumda tipik olarak görülen (konjenital)
yaygın bir genetik bozukluktur. Bozukluk, aralık ve şiddet açısından büyük
ölçüde değişen geniş bir semptom yelpazesi ve fiziksel özellik ile
karakterizedir. Etkilenen birçok kişide, ilişkili anormallikler ayırt edici bir
yüz görünümü içerir; geniş veya perdeli bir boyun; düşük bir posterior saç
çizgisi; tipik bir göğüs deformitesi ve kısa boy. Baş ve yüz (kraniyofasiyal)
bölgesinin karakteristik anormallikleri, genişçe ayarlanmış gözleri (oküler
hipertelorizm); gözlerin iç köşelerini kaplayabilen cilt kıvrımları (efsanevi
kıvrımlar); üst göz kapaklarının sarkması (pitoz); küçük bir çene (mikrognati);
depresif bir burun kökü; geniş tabanlı kısa bir burun; ve düşük yerleşimli,
arkaya döndürülmüş kulaklar (pinnae). Göğüs kemiği (sternum) anormallikleri,
omurganın eğriliği (kifoz ve / veya skolyoz) ve dirseklerin dışa doğru sapması
(cubitus valgus) gibi belirgin iskelet malformasyonları da tipik olarak
mevcuttur. Noonan sendromlu birçok bebekte ayrıca, kalbin sağ alt odasından
akciğerlere (pulmoner kapak darlığı) uygun kan akışının engellenmesi gibi kalp
(kardiyak) kusurları vardır. Ek anormallikler, belirli kan ve lenf damarlarının
malformasyonlarını, kan pıhtılaşmasını ve trombosit eksikliklerini, öğrenme
güçlüklerini veya hafif zihinsel özürlülüğü, testislerin etkilenen erkeklerde
yaşamın ilk yılına kadar inmesini (ve kriptorşidizmi) içerebilir ve / veya
diğer semptom ve bulgular. Noonan sendromu, rasopati yolunu oluşturan çoklu
tekli genlerdeki anormalliklerin (mutasyonlar) neden olduğu otozomal dominant
bir genetik bozukluktur. Noonan sendromu ile ilişkili bazı semptomlar, Turner
sendromu olanlara yüzeysel olarak benzeyebilir (kısa boy, perdeli boyun vb.
Gibi her iki bozuklukla ilişkili olabilecek bazı bulgular nedeniyle). Sonuç
olarak, geçmişte, Noonan sendromu “erkek Turner sendromu”, “kadın yalancı
Turner sendromu” veya “normal kromozomlu karyotipli Turner fenotipi” olarak
anılmıştır. Bununla birlikte, iki bozukluk arasında birçok önemli fark vardır.
Noonan sendromu hem erkekleri hem de kadınları etkiler ve normal bir kromozomal
makyaj (karyotip) vardır. X kromozomunu etkileyen anormallikler ile karakterize
edilen Turner sendromundan sadece kadınlar etkilenir.
Teşhis
Ayrıntılı bir hasta öyküsü,
kapsamlı bir klinik değerlendirme ve çeşitli özel testler. Büyüme eksikliği
veya nedeni bilinmeyen kısa boylu kızlarda Turner sendromundan
şüphelenilmelidir.
Turner sendromu tanısı
genellikle karyotip belirlenerek elde edilen kromozomal analizle doğrulanır.
Karyotipleme, kromozomların sayısını ve yapısını değerlendiren bir laboratuvar
testidir.
Bazı durumlarda, fetal
ultrasonda Turner sendromu ile ilişkili bazı fiziksel bulgular görülebilir.
Örneğin, gelişmekte olan bir fetüsün boynuna yakın lenf sıvısı birikimi bazen
rutin bir fetal ultrasonda görülebilir. Kesin test CVS veya amniyosentez ile
yapılabilir. CVS, gebeliğin 10-12. haftasında yapılır ve doku örneklerinin
plasentanın bir kısmından çıkarılmasını içerirken, amniyosentez 16-18. gebelik
haftasında yapılır ve fetüsün etrafında küçük bir sıvı örneği almayı içerir.
Etkilenen bireyleri
karaciğer, böbrek veya kalp anormallikleri gibi Turner sendromuyla ilişkili
potansiyel semptomların varlığı açısından değerlendirmek için manyetik rezonans
görüntüleme (MRI) gibi spesifik görüntüleme teknikleri uygulanabilir.
Tiroid ve karaciğer
fonksiyonu, kemik yaşı ve büyüme hakkında ek değerlendirme yapılmalıdır.
Hipertansiyon taraması da yapılmalıdır. Doğumda teşhis edilen bebeklere işitme
muayenesi de dahil olmak üzere tam bir kulak, burun ve boğaz muayenesi
yapılmalıdır. Çocuklar, özellikle tekrarlayan orta kulak iltihabı yaşayanlar ve
yetişkinler periyodik işitme değerlendirmesi gerektirir.
Tedavi
Turner sendromunun tedavisi,
her bir bireyde belirgin olan spesifik semptomlara yöneliktir. Tedavi, bir
uzman ekibinin koordineli çabalarını gerektirebilir. Çocuk doktorları, çocuk
uzmanları, cerrahlar, kardiyologlar, endokrinologlar, konuşma patologları,
kulak burun boğaz uzmanları, göz doktorları, psikologlar ve diğer sağlık
uzmanlarının sistematik ve kapsamlı bir şekilde çocuğun tedavisini etkilemesi
gerekebilir. Etkilenen bireyler ve aileleri için genetik danışmanlık önerilir.
Belirli ilaç rejimlerinin ve
/ veya diğer tedavilerin kullanımına ilişkin kararlar, hekim ve sağlık ekibinin
diğer üyeleri tarafından, sendromun özelliklerine dayanarak hastayla dikkatli
konsültasyonda verilmelidir; olası yan etkiler ve uzun vadeli etkiler de dahil
olmak üzere potansiyel faydalar ve riskler hakkında kapsamlı bir tartışma
yapılıp; hasta tercihi ve diğer uygun faktörler değerlendirilmelidir.
Turner sendromu için bir
tedavi yoktur, ancak fiziksel gelişimi artıracak tedaviler geliştirilmiştir.
Uygun tıbbi bakım ile Turner sendromlu kadınlar tam, üretken yaşamlar
sürdürebilmelidir. Etkilenen bireyler için birincil tedaviler büyüme hormonu tedavisi
ve östrojen tedavisidir. Çocuklardaki büyüme hormonu tedavisi Çocuk
endokrinoloji hekimlerince uygun görülen sürede planlanır.
Hormon replasman tedavisi genellikle 12-14
yaşlarında başlar. Çoğu ortalama kız ergenliğe girecek. Ergenliğin
başlatılmasının zamanlaması, büyüme hormonu replasmanındaki büyüme ilerlemesini
de dikkate alır. Bu özellikleri korumak için replasman tedavisine devam
edilmelidir ve çoğu kadın menopoza kadar östrojen ve progesteron tedavisine
ihtiyaç duyacaktır.
Ek tedavi semptomatik ve
destekleyicidir. Örneğin, tiroid hormonu replasman tedavisi tiroid hastalığı
olan bireyleri tedavi etmek için kullanılabilir. İşitme kaybının işitme
cihazlarıyla düzeltilmesi, öğrenme ve sosyal etkileşime yardımcı olabilecek bir
diğer önemli müdahaledir.
Turner sendromlu çocukların
potansiyellerine ulaşmalarını sağlamak için erken müdahale önemlidir.
Tıp uzmanlarının ve iyi bir
sosyal destek sisteminin yardımıyla, TS’li bir kadın tatmin edici, sağlıklı bir
yaşam sürmeyi bekleyebilir.
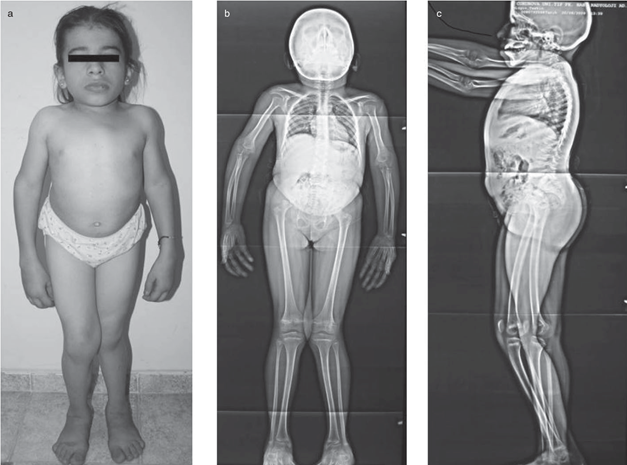
Schimke immüno-ossöz displazisi , kısa boy, böbrek hastalığı ve zayıf bir bağışıklık sistemi ile karakterize bir durumdur. Bu durumu olan insanlarda, kısa boy düzleşmiş omurga kemiklerinden ( vertebralar) kaynaklanır.), Görsel 1kısaltılmış boyun ve gövde ile sonuçlanır. Yetişkin yüksekliği tipik olarak 3 ila 5 feet arasındadır. Böbrek (böbrek) hastalığı sıklıkla hayatı tehdit eden böbrek yetmezliğine ve son dönem böbrek hastalığına (ESRD) yol açar. Etkilenen bireyler ayrıca T hücreleri adı verilen bazı bağışıklık sistemi hücrelerinin yetersizliğine de sahiptir.. T hücreleri yabancı maddeleri tanımlar ve vücudu enfeksiyonlara karşı korur. T hücrelerinin yetersizliği, bir kişinin hastalığa karşı daha duyarlı olmasına neden olur.
Bu rahatsızlığı olan
kişilerde sık görülen diğer özellikler arasında alt sırtın abartılı bir
eğriliği vardır ( lordoz); tipik olarak
göğüste ve sırtta koyulaşmış cilt lekeleri (hiperpigmentasyon); ve burnun
yuvarlatılmış ucu olan geniş bir burun köprüsü.
Schimke immüno-ossöz displazinin daha az görülen belirtileri ve semptomları ,
atardamarların astarında ( ateroskleroz) yağ
birikintileri ve skar benzeri bir doku birikimini içerir.) beyine kan akışını
azalttı (beyin iskemisi), migren benzeri baş ağrıları, yetersiz tiroit bezi (hipotiroidizm), azalmış beyaz kan hücreleri (lenfopeni), az
gelişmiş kalça kemikleri (hipoplastik pelvis), anormal derecede küçük baş
büyüklüğü ( mikrosefali)),
erkeklerde sperm eksikliği (azospermi) ve kadınlarda düzensiz adet kanaması.
Şiddetli vakalarda, doğumda Schimke immüno-ossöz displazinin birçok
belirtisi mevcut olabilir. Bu hastalığın hafif vakaları olan kişiler
geç çocukluğa kadar belirti veya semptom geliştirmeyebilir.
Schimke immuno-ossöz displazili hastaların yaklaşık yarısında SMARCAL1 geninde mutasyon
tanımlanmamıştır. Bu gibi durumlarda, hastalığın nedeni bilinmemektedir.
Bazı insanlar erken çocukluk döneminde şiddetli bir form
geliştirirken, diğerleri çocuklukta veya sonrasında hafif bir form
geliştirir. Kısa boy, omurganın anormal gelişimini ve uzun kemiklerin
uçlarını içeren spondiloepifizeal displaziden kaynaklanır . Neredeyse SIOD’lu
tüm kişilerin böbrek hastalığı vardır, bu da böbrek hastalığına son aşamada ilerler.
Schimke immünoosöz displazisi (SIOD) şiddetine göre değişmektedir. Erken
başlangıç formuna sahip olanlar genellikle ciddi semptomlara ve yaklaşık 9
yıllık ortalama ömre sahiptir. Ölüm nedenleri inme, ciddi fırsatçı
enfeksiyon, kemik iliği yetmezliği, böbrek yetmezliği komplikasyonları,
konjestif kalp yetmezliği veya akciğer hastalığını içerebilir. Daha hafif
belirtileri olanlarda yetişkinlik çağında hayatta kalırlar, böbrek hastalığı
iyi yönetilerek. Bununla birlikte, başlangıç şiddeti ve yaşının mutlaka
yaşam beklentisini kesin göstermeyeceğini not etmek önemlidir, çünkü şiddetli,
erken başlangıçlı SIOD’lu bazı insanlar 20’li ve 30’lu yaşlarında hayatta
kalmıştır.
Genetik Değişiklikler /Etken Faktörler
SMARCAL1 genindeki mutasyonlar, Schimke immüno-osöz displazisi riskini
arttırır. SMARCAL1 gen olan spesifik fonksiyonu bilinmeyen bir protein üretmek için
talimatlar sağlar. SMARCAL1
proteini kromatine bağlanabilir
DNA’yı kromozomlara paketleyen DNA ve protein kompleksidir. Benzer
proteinlerin fonksiyonuna dayanarak, SMARCAL1’in, kromatin remodeling olarak
bilinen bir işlemle diğer genlerin aktivitesini (ekspresyonunu) etkilediği
düşünülmektedir. Kromatinin yapısı, DNA’nın ne kadar sıkı bir şekilde
paketlendiğini değiştirmek için değiştirilebilir (yeniden
yapılandırılabilir). Kromatin remodeling, gelişim sırasında regresyonun
düzenlenmesinin bir yoludur. DNA sıkıca paketlendiğinde, gen ekspresyonu,
DNA’nın gevşek bir şekilde paketlendiğinden daha düşüktür.
SMARCAL1 genindeki mutasyonların, protein
aktivitesini, protein stabilitesini veya proteinin kromatine bağlanma
yeteneğini etkileyerek hastalığa yol açtığı düşünülmektedir. SMARCAL1 genindeki mutasyonların kromatin
remodelingine ve diğer genlerin ekspresyonuna müdahale edip etmediği açık değildir.
Schimke immüno-disöz displazisi ile ilişkili mutasyonlar, SMARCAL1 proteininin olağan
fonksiyonlarını bozar veya herhangi bir fonksiyonel proteinin üretimini
önler. Fonksiyonel bir protein eksikliğine neden olan mutasyonlara sahip
olan insanlar, aktif fakat hatalı çalışan bir proteine yol açan mutasyonlara
sahip olanlardan daha şiddetli bir şekilde bu hastalığa sahip olma
eğilimindedir. Bununla birlikte, SMARCAL1 gen
mutasyonlarına sahip kişilerin Schimke immüno-kemikli displazisi geliştirmesi
için , şu anda bilinmeyen diğer genetik veya çevresel faktörlerin de
bulunması gerekir.
Belirti ve Semptomlar
Schimke
immünoosöz displazisi (SIOD) vücudun birçok bölümünü etkiler. Yavaş büyüme
genellikle SIOD’un ilk işaretidir. İnsanlar genellikle kısa bir boyun ve
gövde geliştirir (orantısız şekilde).kısa
boy) spondiloepifizeal displazi nedeniyle . Kemik
anormallikleri tipik olarak omurga veya kalçalarda gelişir. SIOD’lu birçok
kişi sonunda kalça
protezi ameliyatına ihtiyaç
duyar .Böbrek hastalığı tanı anında var olur veya birkaç
yıl içinde gelişir ve sonuçta böbrek yetmezliğine ilerler . Neredeyse
tüm SIOD’lu kişilerin kanı varhücreEn sık T-hücrelerinin
eksikliği. Bu, hayati tehlike oluşturabilecek enfeksiyon riskinin
artmasına neden olur.
Schimke immuno-osseöz displazisi çok nadir görülen bir
durumdur. Kuzey Amerika’da yaygınlığın 1 milyondan 3 milyona kadar bir
insan olduğu tahmin edilmektedir.
Kalıtım Paterni\Deseni
SMARCAL1 genindeki mutasyonlar, otozomal resesif paternde kalıtsaldır bu, Schimke
immüno-ossöz displazisi riskinin artmasının,
her hücrede SMARCAL1 geninin
her iki kopyasındaki mutasyonlardan kaynaklandığı anlamına
gelir. Otozomal resesif hastalığı olan bir bireyin ebeveynlerinin her biri
mutasyona uğramış genin bir kopyasını taşır, ancak bunlar genellikle durumun
belirtilerini ve semptomlarını göstermezler.
Kavramsal olarak, etkilenen
bir bireyin her
birinin % 25’i etkilenme şansı, %50’sinin asemptomatik bir taşıyıcı olma olasılığı ve % 25’inin de
etkilenmeme ve taşıyıcı olmama olasılığı vardır. Ailede patojenik
varyantların her ikisi de biliniyorsa taşıyıcı testi ve doğum öncesi testi
mümkündür.
Teşhis Yöntemleri ve Tedavileri
Teşhis:
Aşağıdaki temel
özelliklere sahip kişilerde SIOD tanısından şüphelenilir:
Tanı klinik muayeneden
sonra konulabilir. Genetik test tespit
etmek mutasyonlariçinde SMARCAL1 genteşhisi
onaylayabilir.
Kıkırdak kılı hipoplazisi (bu terime bakınız) ana
ayırıcı tanıdır.
Tedavi:
Schimke
immunoosseöz displazinin (SIOD) tedavisi her bireyin şiddetine ve bireysel
semptomlarına bağlıdır. Kalça, böbreklerin düzenli olarak izlenmesi,bağışıklık sistemive kan tavsiye edilir.
SIOD için gerekli olabilecek tedavi örnekleri
şunları içerir:
Böbrek diyaliz veya
nakli
Kalça protezi
Nötropeninin
granülosit ile tedavisi koloni uyarıcı faktör veya
granülosit-makrofaj koloni uyarıcı faktör
Kemik iliği
nakli immün yetmezlik için (transplantasyon sonrası birkaç ölüm
bildirilmiş olmasına rağmen)
Otoimmün semptomları
olanlar için bağışıklık sistemini baskılayan ilaçlar
Şiddetli
papilloma tedavisinde Imiquimod ve cidofovir (bir
antiviral)virüs cilt enfeksiyonları
Geçici iskemik
atakları (“mini vuruş”) veya felç tedavisinde kan akışını iyileştiren
veya kanın pıhtılaşmasını azaltan ajanlar
Hipotiroidizmin standart
tedavisi
Standart
tedavi skolyoz
Hastalıkla İlişkili Genler
Schimke immüno-ossöz displazisi (SIOD),
spondiloepifizeal displazi ve orantısız kısa boy, yüz dismorfizmi, T hücreli
immün yetmezlik ve nefrotik sendromlu glomerülonefrit ile karakterize multisistemik
bir hastalıktır.
SIOD , SMATCAL1 genindeki (2q35), kromatin remodeling proteini hHARP’yi
(ayrıca kromatin alt-ailesi A-benzeri protein 1’in SWI / SNF ile ilgili
matris-ilişkili aktin bağımlı regülatörü olarak da bilinir)
kodlayan mutasyonlardan kaynaklanır .
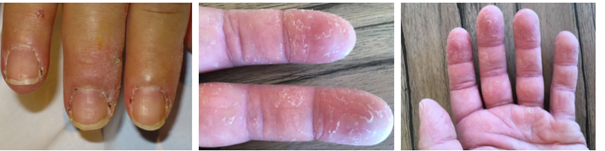
Antisynthetase sendromu; kronik bir otoimmün durumudur. Vücudun kaslarını ve çeşitli kısımlarını etkiler. Birçok belirti ve semptomu kapsar. Kas iltihabı (miyozit), poliartrit (birçok eklem iltihabı), interstisyel akciğer iltihabı, ellerin kalınlaşması ve çatlaması ve Raynaud fenomenini kapsayana karakterize bir durumdur. Esase vakanın altında yatan neden bilinmemektedir ancak vücutta biyosentezi olan aminoasil-tRNA sentetazları adındaki enzimlere saldıran otoantikoların (normal antikorlara saldıran antikorlar) üretiminin, sendromun nedeni olarak düşünülen kanılardan birisidir. Bu otoantikorlar viral enfeksiyonla beraber ortaya çıkabilir veya viral enfeksiyondan sonra da ortaya çıkabilir. Hastaların genetik yatkınlığı da söz konusu olabilir. Tedavi, her insanda belirti ve semptomlara dayanmaktadır; kortikosteroidleri, immünosüpresif ilaçlar veya fizik tedaviyi barındırabilir.
Antisynthetase sendrom hastalarının uzun süre bakımı, kronik immünosüpresif tedavinin olumsuz etkilerine ve kompilasyonlarına maruz kalabilir. Pulmoner hipertansiyon, malignite ve azalmış sağkalımı gerektiren ilerleyici interstisyel akciğer hastalığını içerebilen hastalıkla ilişkili doku bozuklukları beraberinde getirir.
Akciğer transplantasyonu, pulmoner hipertansiyon, malignite ve azalmış sağkalımı gerektiren doku bozukluğu olan interstisyel akciğer hastalığının klinik özellikleri hakkında daha fazla tanı, araştırma, tanı, teşhis, tedavi ve daha fazla farkındalığın oluşumu Antisynthetase sendrom hastaları için umud vaad edilmektedir.
Antisynthetase Sendrom Semptomları:
Kas
iltihabı (miyozit)
Poliartrit
(bi kaç eklemde iltihap)
Ateş
Mekanik
eller
Raynaud
Fenomeni
İnterstisyel
akciğer hastalığı (spesfik olmayan akciğer iltihabı)
Başlıca
semptomlar bunlarla beraberinde olup zamanla değişme ihtimali de vardır.
Antisynthetase Sendromu Tanısı Nasıl Yapılır?
Kas enzimleri, örneğin kreatinin
kinaz (CK) ve aldolaz: bunlar genellikle yükselir
Kas antikorları
Elektromiyografi (EMG)
Etkilenen kasların manyetik
rezonans görüntüleme ( MRI )
Kas biyopsisi
Akciğer fonksiyon testleri
Göğsün yüksek
çözünürlüklü bilgisayarlı tomografi taraması ( BT )
Yutma güçlükleri ve aspirasyon
riskinin değerlendirilmesi
Akciğer biyopsisi
Antisintaztaz Sendromu Nasıl Tedavi Edilir?
Glukokortikosteroidler antisynthetase
sendromu için tedavinin temel dayanağıdır ve genellikle birkaç ay veya yıl
boyunca gereklidir. Prednizon başlangıçta 4-6 hafta boyunca yüksek
dozlarda (1 mg / kg / gün) hastalık kontrolünü sağlamak için verilir,
daha sonra remisyonu sürdürmek için en düşük etkili doza 9-12 ay
boyunca yavaşça azaltılır . Daha ciddi vakalarda, 3-5 gün
boyunca darbeli intravenöz (IV) metilprednizolon gerekebilir.
Steroid kaynaklı osteoporoza ve Pneumocystis jirovecii gibi bazı mantar enfeksiyonlarına karşı profilaktik tedavi önerilir . Tedaviye başlamadan önce aşılama ihtiyacı değerlendirilmelidir.
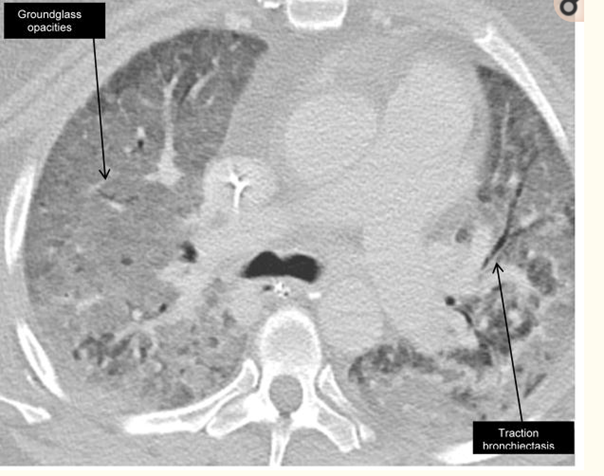
Şekil 1) Antisynthetase sendrom yüksek çözünürlüklü BT’si.
The Diagnosis and Treatment of Antisynthetase Syndrome Leah J. Witt, MD,1 James J. Curran, MD,2 and Mary E. Strek, MD1 PMCID: PMC500639 NIHMSID: NIHMS80052PMID: 27594777
Konjenital
alfa2 antiplazmin eksikliği,
küçük travma ve spontan kanama epizodlarını takiben uzun süreli kanama ve
ekimozların neden olduğu konjenital alfa2 antiplazmin eksikliğinin neden olduğu
nadir bir hemorajik bozukluktur (genellikle uzun kemiklerin diyafizi gibi
alışılmadık yerlerde). Konjenital alfa2 antiplazmin eksikliği otozomal
resesif bir şekilde kalıtsaldır.
Konjenital alfa2 antiplazmin
eksikliği, ilk olarak 1969 yılında Japonya’nın güneybatısındaki Okinawa
adasında yaşayan bir çocukta Masateru Kohakura tarafından
tanımlanmıştır. Çocuğun kesiklerden uzun süreli kanama, subkutan kanamalar
ve travmatik eklem kanaması gibi hemorajik diyatezi vardı. Daha sonra, tüm
soyağacı, ortak atalardan gelen aile üyeleri arasında ve hepsi adada yaşayan 3
akraba evliliği ile tanımlandı. alfa 2-antiplazmin, insan dolaşımda plazmin
doğal inhibitörüdür ve fibrinoliz düzenlenmesinde önemli bir rol
oynar. Plazmin-antiplazmin sistemi, fibrin polimerlerinin çözünür
fragmanlara çözülmesini düzenler.
Genetik Değişiklik/Etken Faktörler
Alfa 2-antiplazmin eksikliği konjenital veya edinsel olabilir. Konjenital alfa 2-antiplazmin eksikliği olan hastalar, plazmin inaktivasyonu ve hemostatik fibrin tıkacının erken lizizine bağlı olarak ciddi bir hemorajik bozukluk ile başvurabilir. Travma veya ameliyattan sonra bazen kanama gecikebilir.
Edinilmiş alfa 2-antiplazmin eksikliği karaciğer hastalığı (sentez azalması), yaygın damar içi pıhtılaşma (artan tüketim), nefrotik sendrom (idrar kaybı), amiloidoz veya trombolitik tedavi sırasında görülebilir.
Alfa-2-plazmin inhibitörü
eksikliğine PLI (SERPINF2; 613168 ) genindeki mutasyonun neden olduğu
vakalarda tespit edilmiştir.
Belirti ve Semptomlar
Homozigot alfa 2-antiplazmin eksikliği olan hastalar göbek kordonundan kanama ile çocuklukta erken ortaya çıkabilir ve ömür boyu kanama eğilimi gösterir.
Kanama belirtileri konjenital
hemofili olan bir hastaya benzer ve aşağıdakileri içerebilir:
Minör travma sonrası gecikmiş kanama başlangıcı
Mukozal kanama dahil olmak üzere kesik ve
yaralardan uzun süreli kanama
Kas kanaması dahil artan morarma ve hematomlar
Travma sonrası spontan eklem kanaması yerine
eklemlere kanama
Ameliyat sonrası aşırı kanama (daha hafif
vakalarda ipucu olabilir)
Aşırı morarma
Steroid olmayan antienflamatuar ilaçların
(NSAID’ler) kullanımından sonra artan kanama
Bademcik ameliyatı, adenoidektomi ve diş çekimi
sonrası uzun süreli kanama bildirilmiştir.
Etkilenen bireylerde spontan
hemotoraks, hematüri, burun kanaması ve kas hematomları da
tanımlanmıştır. Uzun kemiklerin diyafizinde olağandışı intramedüller
kanaması olan birçok hasta, kendiliğinden veya travma ile ortaya
çıkmıştır.
Heterozigot alfa 2-antiplazmin eksikliği daha hafif kanama epizodları ile ilişkili olabilir ve çoğu hasta asemptomatik olabilir. Diş veya cerrahi prosedürlerden sonra uzun süreli kanama meydana gelebilir. Alfa 2-antiplazmin seviyelerinin düşmesinin bir sonucu olarak, ilerleyen yaşla birlikte kanama belirtileri ortaya çıkabilir.
Genetik Görülme Sıklığı
Alfa 2-antiplazmin eksikliği
dünya çapında tarif edilen sadece bir avuç vaka ile çok nadir görülen bir
hastalıktır. Sonuç olarak, bu bozukluğun yaygınlığı ve etnik tercihleri
bilinmemektedir. Yüksek verimli genomik testlerin daha yaygın kullanımı
ile, bu durumun sıklığı hakkında daha fazla bilgi elde edilebilir.
Kalıtım Paterni/Deseni
Alfa 2-antiplazmin konjenital
eksikliği, otozomal resesif bir kalıtım paternini takip eder. Homozigot
eksikliği olan hastaların ailelerinde akraba evliliği öyküsü vardır.
Birkaç alellik varyant
tanımlanmıştır.
Teşhis Yöntemleri ve Tedavileri
Teşhis
Alfa 2-antiplazmin eksikliği
için spesifik bir tahlil, bir hastanın olağan tarama tahlilleri ile
tanımlanamayan bir kanama diyatezi olduğunda yapılır. Hem alfa
2-antiplazmin antijeni hem de aktivite seviyeleri ölçülebilir.
İki tür alfa 2-antiplazmin
eksikliği vardır:
Tip 1 – Azalan antijen ve aktivite seviyeleri
Tip 2 – Normal antijen seviyesi ile düşük
aktivite seviyesi
Homozigot hastalar genellikle
saptanamayan seviyelere sahipken heterozigot hastalar normal seviyelerin% 40 -%
60’ına sahiptir. Normal plazma konsantrasyonu 0.7 mg / L’dir.
Tedavi
Alfa 2-antiplazmin ile
ilişkili kanama genellikle aminokaproik asit veya traneksamik asit gibi
antifibrinolitik ajanlarla etkili bir şekilde tedavi edilir. Bu ajanlar
herhangi bir cerrahi veya diş prosedüründen önce kanamaya yanıt olarak veya
profilaksi olarak kullanılabilir. Antifibrinolitik ajanlar esas olarak
plazminojenin fibrine bağlanmasını önleyerek, endojen fibrinolizi inhibe ederek
ve hemostatik tıkacı stabilize ederek etki gösterir. Oral veya intravenöz
olarak verilebilirler ve genellikle iyi tolere edilirler.
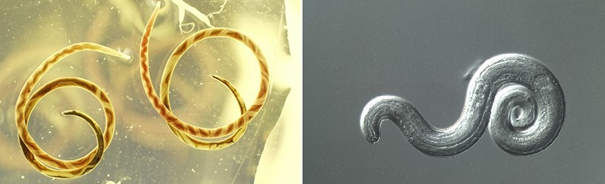
Strongyloidiasis , yuvarlak kurt Strongyloides
stercoralis’in (S. stercoralis) neden olduğu parazitik bir hastalıktır.
İnsanlar, solucanlarla kontamine olmuş toprakla temas ettiklerinde enfeksiyonu
yakalarlar. Sıklıkla semptom
görülmezken, karın ağrısı, öksürük, ishal, döküntü, nedensiz kilo kaybı ve
kusma görülebilir. Enfeksiyon, ivermektin gibi anti-solucan ilaçlarıyla tedavi
edilir . Strongiloides tropikal ve alt tropikal bölgelerde bulunur, aynı
zamanda güney ABD dahil ılıman bölgelerde bulunabilir.
Klinik Tanım
Akut enfeksiyon, serpiginous ürtikeryal döküntü, öksürük, nefes darlığı, gastrointestinal semptomlar (ağrı ve yumuşak dışkı dahil) ve kutanöz, alerjik belirtiler ile karakterizedir; ancak enfekte olmuş kişilerin yarısından fazlası asemptomatik kalmaktadır. HTLV1 enfeksiyonu gibi alta yatan başka hastalığı olan veya kortikosteroidler veya immünosüpresif tedavi gören hastalarda Strongyloid hiperinfeksiyon sendromu (SHS) ortaya çıkabilir ve sıklıkla sepsis, şok ve akut solunum sıkıntısı sendromu ile sonuçlanır. Solunum, gastrointestinal, kutanöz ve nörolojik semptomlar değişken yoğunluklarda görülür, ancak SHS’nin ayırt edici özelliği organ yetmezliğinin şiddetidir.
Belirtiler
Akut ve kronik strongyloidiasis asemptomatik
olabilir. Akut strongiloidiazisinin , ilk belirtisi larvanın cilde girdiği
yerinde bir kaşıntı, eritemli döküntü olabilir. Larvalar akciğerlerden ve
trakeadan göç ettikçe öksürük gelişebilir. Gastrointestinal sistemdeki larvalar
ve yetişkin solucanlar karın ağrısı, ishal ve anoreksiye neden olabilir.
Kronik strongyloidiasis , otoenfeksiyon
nedeniyle yıllarca sürebilir. Asemptomatik olabilir veya gastrointestinal,
pulmoner ve / veya kutanöz semptomlarla karakterize edilebilir.
Gastrointestinal şikayetler karın ağrısı ve aralıklı ishal ve kabızlığı içerir.
Açık gastrointestinal kanama oluşabilir ve nadiren dışkıda kan testleri pozitif
olabilir. Semptomlar ülseratif kolit , diğer kronik malabsorpsiyon nedenleri
veya duodenal tıkanıklığın semptomları ile devam edebilir .Ağır enfeksiyonlarda,
otoenfekte larvalar akciğerlerden geçerken öksürük, hırıltılı solunum ve
eozinofili ile Löffler sendromuna neden olabilmesine rağmen, akciğer
semptomları nadirdir . Semptomlar alerjik astım veya kronik obstrüktif akciğer
hastalığına (KOAH) yönelebilir.
Patofizyolojisi
Strongyloides erişkin kurtları, duodenum ve
jejunumun mukozasında ve submukozasında yaşar. Serbest bırakılan yumurtalar,
rabditiform larvaları serbest bırakarak bağırsak lümeninde yumurtadan çıkarlar.
Larvaların çoğu dışkıda atılır. Toprakta birkaç gün sonra, bulaşıcı filariform
larvalara dönüşürler. Kıl kurtları gibi , Strongyloides larvaları insan derisinden
girebilir, kan dolaşımı yoluyla, kılcal damarlardan geçerek, solunum yollarından
akciğerlere ulaşabilir, yutulabilir ve yaklaşık 2 hafta içinde olgunlaşarak
bağırsağa ulaşır. Toprakta, insanlarla temas etmeyen larvalar, larvaları insan
bir konakçıya tekrar girmeden önce birkaç nesil üreyebilen serbest yaşayan yetişkin
solucanlara olgunlaşır.
Epidemiyolojisi
Dünya çapında 30 ila 60 milyon insanı
etkilemektedir ve Afrika, Batı Hint Adaları, Orta ve Güney Amerika, Hint
Okyanusu bölgesi, Güney Doğu Asya gibi alt tropikal bölgelere endemiktir.
Etolojisi
2.5 mm uzunluğunda olan dişi nematodlar,
insanların ince bağırsağında yaşar. Yumurtalar, larvaların dışkılarla atılması
için ince bağırsağın kapağına yerleştirilir. Nemli zeminde, bu larvalar
bulaşıcı aşamalarına doğrudan veya cinsel üreme aşamasından sonra ulaşır. Bu
bulaşıcı formda cilde doğrudan nüfuz edebilirler. Bulaşıcı aşamaya doğru evrim,
bazı durumlarda gözlenen uzun parazitoz süresini (30 yıldan fazla) açıklayan
sindirim sistemi içinde de gerçekleşebilir.
Teşhis
Dışkı veya duodenal dahil örneklerin mikroskobik incelemesi ve
hiperinfeksiyon sendromunda bronşiyal yıkama, balgam veya diğer vücut
sıvılarıyla larvaların tanımlanması
Antikorlar için enzim-immüno analizi
Tek bir dışkı örneğinin mikroskobik
incelemesi, komplike olmayan Strongyloides enfeksiyonlarının yaklaşık% 25’inde
larvaları tespit eder . Konsantre dışkı örneklerinin tekrar tekrar incelenmesi
hassasiyeti artırır; en az 3 ve en fazla 7 dışkı örneği önerilir. Özel dışkı
inceleme yöntemleri hassasiyeti arttırır. Bunlar arasında besin agar plaka
kültürü, Baermann huni tekniği ve Harada-Mori filtre kağıdı tekniği
bulunmaktadır.
Hiperinfeksiyon sendromunda filariform larvalar
dışkı, duodenal içerisinde, balgam ve bronşiyal yıkamalarda ve nadiren beyin
omurilik sıvısında (BOS), idrar, plevral veya asit sıvısında bulunabilir.
Akciğer dokusunun biyopsilerinde veya diğer organların dokularında da görülebilirler.
Akciğer x-rayleri yaygın interstisyel infiltratlar, konsolidasyon veya apse
gösterebilir.
Serumdaki anti-strongiloid antikorlarını
tanımlamak için birkaç bağışıklık tanısı testi mevcuttur. Daha büyük
hassasiyeti ( > % 90) nedeniyle enzim immünolojik testi (EIA) önerilir .
Serum IgG antikorları yaygın yayılmış strongiloidiazisi olan immün sistemi
baskılanmış hastalarda bile tespit edilebilir, ancak saptanabilir antikorların
yokluğu enfeksiyonu dışlamaz. Filariasis veya diğer nematod enfeksiyonları olan
hastalarda çapraz reaksiyonlar yanlış pozitif testlere neden olabilir. Antikor
testi sonuçları, anlık enfeksiyonu geçirilmiş enfeksiyondan ayırt etmek için
kullanılamaz. Pozitif bir test, parazitolojik tanı koymaya yönelik çabaların
sürdürülmesini gerektirir.
Serolojik izleme faydalı olabilir çünkü
başarılı kemoterapiden sonraki 6 ay içinde antikor seviyeleri düşer.
S. stercoralis tanısı için polimeraz zincir
reaksiyonu (PCR) tabanlı yöntemler geliştirilmektedir.
Eozinofili sıklıkla bulunur, ancak
kortikosteroidler veya sitotoksik kemoterapötik ilaçlar gibi ilaçlar tarafından
bastırılabilir.
Tedavi
Ivermektin
Alternatif olarak, albendazol
Strongiloidiazisi olan tüm hastalar tedavi
edilmelidir. İvermektin ile tedavi oranı albendazole göre daha yüksektir.
Ivermectin 200 mcg / kg, 2 gün boyunca günde
bir kez oral olarak, karmaşık olmayan enfeksiyon için kullanılır ve genellikle
iyi tolere edilir. İvermektin ile tedaviden önce hastalar, Loa Loa endemiğinin
bulunduğu Afrika’nın merkez bölgelerine seyahat etmişlerse, Loa Loa tanı testi
uygulamalıdır çünkü ivermektin , loiasis ve yüksek mikrofilaryal hastalarda
ciddi reaksiyonlara neden olabilir. Albendazol 400 mg, 7 gün boyunca günde iki
kez oral yoldan, kuvvetliloidiazisin tedavisi için bir alternatiftir.
İmmün sistemi baskılanmış hastalar balgam ve /
veya dışkı 2 hafta boyunca negatif olana kadar uzun süreli tedavi gerektirir.
Bazen tekrarlanan tedavilere ihtiyaç vardır. Oral ilaç verilemeyen ağır
hastalarda, ivermektinin rektal preparatları veya veteriner deri altı formülasyon
ivermektini kullanılmıştır.
Strongiloidiazili hastalarda hiperinfeksiyon
sendromu yaşamı tehdit eden bir tıbbi acil durumdur. İbitmektin 200 mcg / kg
oral olarak günde bir kez rahabditiform ve filariform larvalar için balgam ve
dışkı incelemeleri 2 hafta boyunca negatif olana kadar devam eder. Geniş
spektrumlu antibiyotikler, bağırsaktan larva istilası ile ilişkili eş zamanlı
polimikrobiyal bakteriyel enfeksiyonları tedavi etmek için kullanılır.
Strongyloidiasis tedavisinden sonra, 2 ila 4
hafta sonra tekrarlanan dışkı muayeneleri ile tedavi belgelenmelidir. Dışkı
pozitif kalırsa, yeniden tedavi belirtilir.
Prognoz
Tedavi edilmezse yaşam boyu enfeksiyon
mümkündür. Çoğu hasta kronik hastalıklarda bile asemptomatik kalır, ancak
prognoz komplikasyonların gelişimine bağlıdır. Yaygın enfeksiyon vakaların%
60-70’inde ölümcüldür.
Anjiostrogylosis,
(sıçan akciğer kurdu enfeksiyonu) endemik olarak görülen zoonotik gıda kaynaklı,
hatta yıkanmamış meyve ve sebzeleri tükettikten sonra ortaya çıkabilen bir
hastalıktır.
Güneydoğu
Asya ve Pasifik Adaları Angiostrongylus pulmoner nematodunun neden olduğu cantonensis.
Enfeksiyon, sebzelerde bulunan bulaşıcı larvaların yutulması veya çiğ ve az
pişmiş salyangozlarda, midye salyangozlarında, kara yengeçlerinde, tatlı su
yengeçlerinde karides, kurbağa ve kertenkele yenilmesiyle ilişkilidir.
Enfeksiyonun ana özelliği eozinofilik menenjittir; ateş, baş ağrısı, halsizlik,
yorgunluk, rinit ile klinik olarak kendini gösteren serebral, bulanık ve çift
görme, öksürük, boyun tutulması, enterit ve kabızlık nematodların
bağırsaklardan akciğerlere, merkezi sisteme hareketinden kaynaklanan
parasteziler sinir sistemi ve gözlere ulaşarak belirtileri başlatır. Şiddetli,
tedavi edilmeyen vakalarda koma ve ölüm meydana gelebilir. Sıklıkla enfeksiyon,
tedavi veya ciddi sonuçlar olmaksızın düzelir, ancak ağır parazit yükü olan
durumlarda, enfeksiyon çok şiddetli olabilir.
Genetik Değişiklikler/Etken Faktörler
A.cantonensis’in birçok
vektörü vardır, en yaygın olanı , Pasifik adalarındaki dev Afrika
kara salyangozu ( Achatina fulica ) ve Tayland ve
Malezya’daki Pila cinsinin salyangozları dahil olmak üzere
birkaç salyangoz türüdür . Altın elma salyangozu A.
canaliculatus , Çin’deki en önemli vektördür. Tatlı su karidesleri,
yengeçler veya diğer paratenik veya taşıma konakları da vektör olarak hareket
edebilir. İnsanlar tesadüfi konakçılardır; larvalar insanlarda çoğalamaz
ve bu nedenle insanlar A. cantonensis yaşam
döngüsüne katkıda bulunmazlar.
Sıçan akciğer kurdu olan nematod
(yuvarlak kurt) Angiostrongylus
cantonensis , insan eozinofilik menenjitinin en yaygın
nedenidir.
Belirti ve Semptomlar
Enfeksiyon ilk önce yavaş
yavaş yükselerek ilerleyen ateş, şiddetli karın ağrısı, bulantı, kusma ve halsizlik,
daha sonra merkezi sinir sistemi (CNS) semptomları ve boynun şiddetli baş
ağrısı ve sertliği ile kendini gösterir. CNS semptomları hafif bilişsel
bozukluk ve yavaş reaksiyonlarla başlar ve çok şiddetli bir formda genellikle
bilinç kaybına ilerler.
Göz istilası belirtileri
arasında görme bozukluğu, ağrı, keratit ve retina
ödemi bulunur . Solucanlar
genellikle ön odada ve vitrözde görülür ve
bazen cerrahi olarak çıkarılabilir.
Kuluçka süresi :
İnsanlarda kuluçka dönemi
enfeksiyondan sonra genellikle 1 haftadan 1 aya kadardır ve 47 güne kadar
uzayabilir. Bu aralık değişir, çünkü insanlar ara konakçıdır ve yaşam
döngüsü bir sıçanda olduğu gibi tahmin edilebilir şekilde devam etmez.
Görsel 1, kaynak: Sol: Sıçan akciğerlerinden iki Angiostrongylus erişkin kadın iyileşti. Her iki solucanda görülen ayırt edici, sarmal desen beyaz uterus tüpleri ve kırmızı, kan dolu bağırsak tarafından yaratılır. Sağ: Angiostrongylus cantonensis üçüncü evre (L3), bir sümüklübeden enfektif larva. Diferansiyel girişim kontrast (DIC) mikroskobu altında yakalanan görüntü.
Genetik Görülme Sıklığı
Genetik
bir bilgi bulunmamaktadır. A. cantonensis ve vektörleri Güneydoğu Asya ve
Pasifik Havzası’na endemiktir. Küreselleşme gittikçe daha fazla yere
yayılmasına ve daha fazla gezgin parazitlerle karşılaştıkça enfeksiyon giderek
daha önemli hale geliyor. Parazitler muhtemelen gemilerde kaçak yol olarak
seyahat eden sıçanlardan ve endemik alanların dışında salyangoz vektörlerinin
sokulmasıyla etkili bir şekilde seyahat ederler.
Parazit
endemik bölgelerin dışında nadiren görülür ve bu durumlarda hastalar genellikle
endemik bir bölgeye seyahat öyküsü vardır.
Angiostrongyliasis’in
teşhisi, angiostrongylus larvalarının kendilerini
gösterme zorluğu nedeniyle karmaşıktır ve genellikle eozinofilik
menenjitin varlığına ve salyangoz konakçılarına maruz kalma öyküsüne dayanarak
yapılır. Eozinofilik menenjit genellikle BOS’ta > 10 eozinofil / μL veya toplam BOS lökosit sayısında en
az% 10 eozinofil içeren bir menenjit olarak karakterize
edilir . Beyin omurilik sıvısında bulunan veya cerrahi olarak gözden
çıkarılan solucanlar Angiostrongyliasis’i teşhis etmek için tanımlanabilir.
Lomber ponksiyon her zaman
menenjit şüphesi olan durumlarda yapılmalıdır. Eozinofilik menenjit
vakalarında, BOS’ta bulunduklarında bile nadiren solucanlar üretecektir, çünkü
sinirlerin sonuna yapışmaya eğilimlidirler.
BT veya MRG’de hem gri hem
de beyaz cevher invazyonlu beyin lezyonları görülebilir. Bununla birlikte,
MRG bulguları sonuçsuz olma eğilimindedir ve genellikle spesifik olmayan
lezyonlar ve ventriküler genişlemeyi içerir. Bazen göç eden solucanlar
tarafından üretilen bir kanama vardır ve teşhis değeri vardır.
Yüksek eozinofilleri olan
hastalarda seroloji, başka bir parazit ile enfeksiyondan ziyade
Angiostrongylias tanısını doğrulamak için kullanılabilir.
En kesin tanı her zaman BOS
veya gözde bulunan larvaların tanımlanmasından kaynaklanır, ancak bu nadirlik
nedeniyle yukarıdaki testlere dayanan bir klinik tanı büyük olasılıkla vardır.
Tedavi
Angiostrongiliyazis için
gelişme aşamasında hiçbir aşı yoktur.
Bireyler için öneriler
Endemik bölgelerde enfeksiyondan
kaçınmak için ;
Salyangoz ve tatlı su karidesleri gibi pişmemiş vektörlerin tüketiminden
kaçının
Vektörlerle kirlenmiş olabilecek açık kaynaklardan içme suyundan kaçının
Küçük çocukların canlı salyangozlarla oynamasını veya yemesini önleyin
Anjiyostrongiliyazis
tedavisi iyi tanımlanmamıştır, ancak çoğu strateji solucanları öldürmek için
anti-parazitiklerin, solucanlar ölürken iltihabı sınırlayan steroidlerin ve
menenjit semptomlarını yönetmek için ağrı kesici ilaçların bir kombinasyonunu
içerir.
Semptomatik tedavi, bulantı,
kusma, baş ağrısı ve bazı durumlarda sinir hasarı veya kas atrofisine bağlı
kronik ağrı gibi belirtiler için endikedir.
İnsanlara yapılan en iyi
öneri, yiyeceğiniz çiğ ürünleri iyice yıkayın, ellerinizi yıkayın, salyangoz
veya sümüklü böcek veya fare yemeyin.
Gorham-Stout hastalığı
(kaybolan kemik hastalığı) (GSD), lenfatik damarların çoğalması ve genişlemesi
ile ilişkili nadir bir masif osteoliz hastalığıdır. GSD vücuttaki herhangi bir
kemiği etkileyebilir ve monostotik veya poliostotik olabilir. Tıp literatüründe
kaybolan kemik hastalığı, masif osteoliz olarak da bilinen Gorham-Stout
hastalığı (GSD), ilerleyici kemik kaybı (osteoliz) ile karakterize nadir bir
kemik bozukluğudur ve lenfatik damarların aşırı çoğalması (proliferasyonu).
Etkilenen bireyler, ilerleyen kemik yıkımı ve emilimine maruz kalırlar. Birden
fazla kemik tutulabilir.
GSD her yaşta ortaya
çıkabilir, ancak genellikle çocuklarda ve genç yetişkinlerde (ortalama 13 yıl)
teşhis edilir. GSD vücuttaki herhangi bir kemiği etkileyebilir, ancak en yaygın
olarak kaburgaları, ardından kafatası, klavikula ve servikal omurga etkiler.
Etkilenen diğer alanlar arasında maksillofasiyal kemikler (çoğunlukla çene
kemiği), sternum, humerus, el, femur ve ayak bulunur. GSD monostotik veya
poliostotik olabilir ve semptomlar etkilenen vücut bölgelerine göre değişir. En
yaygın semptom lokalize ağrıdır. Etkilenen uzuvların şişmesi, zayıflığı ve
fonksiyonel bozukluğu da fark edilir. Dentoalveoler bölgede hareketli dişler,
malokluzyon, mandibular sapma ve kemik deformitesi görülebilir. Torasik
tutulumu olan hastalar solunum sıkıntısı (şilotoraksın neden olduğu)
gösterebilir. Vertebral tutuluma sekonder şiddetli nörolojik defektler ve felç
de gözlenir. Servikal omurga veya kafatasının hastalığı olan hastalarda beyin
omurilik sıvısı sızıntısı gelişebilir. GSD, bir kemik kırılmasından sonra
(spontan veya küçük travmadan sonra) bulunabilir.
GSD etiyolojisi hala anlaşılması güçtür.
Patolojik süreç, kemiğe bitişik veya kemik içindeki endotel kanallarının iyi
huylu vasküler proliferasyonu olup kemik trabekülünün aşırı incelmesine,
osteoklast aracılı rezorpsiyona ve kemiğin fibröz doku ile değiştirilmesine yol
açar. Doku örnekleri lenfatik endotel hücre markerleri için pozitif test eder,
bu da GSD’nin düzensiz lenfanjiyogenez içeren bir hastalık olduğunu düşündürür.
Kemik kaybı (osteoliz) enfeksiyon,
inflamasyon, kanser ve bazı endokrin bozuklukları içeren çeşitli farklı
durumlardan kaynaklanabilir. GSD’nin ayırıcı tanısında Hajdu-Cheney sendromu,
Paget hastalığı, romatoid artrit, fibröz displazi, Langerhans hücre
histiyositozu, Winchester sendromu, karpal tarsal osteoliz, idiyopatik
multisentrik osteoliz, nefropatili multisentrik osteoliz ve eozopozizofizozit
yer alır . (Bu bozukluklar hakkında daha fazla bilgi için, Nadir Hastalık
Veritabanında arama teriminiz olarak belirli bir bozukluk adı seçin.)
Genelleştirilmiş lenfatik anomali (daha
önce lenfanjiyotoz olarak bilinen GLA) GSD ile yakından ilişkilidir. GLA
hastaları multifokal lenfatik malformasyonlara sahiptir. Bu malformasyonlar
kemikte mevcut olabilir, ancak GSD’de görüldüğü gibi kortikal kemik kaybına
neden olmaz.
Belirti ve Semptomlar
Gorham’ın yok olan kemik
hastalığı ve fantom kemik hastalığı olarak da bilinen Gorham hastalığı
(belirgin GOR-amz), içerisindeki bozuk, ince duvarlı vasküler veya lenfatik
kanalların kontrolsüz çoğalmasıyla karakterize, çok nadir bir iskelet
durumudur. kemiğin emilimine ve kemiğin anjiyomlar ve / veya fibroz ile değiştirilmesine
yol açan kemik hastalığı. GSD’den yaygın olarak etkilenen bölgeler arasında
kaburgalar, omurga, pelvis, kafatası, köprücük kemiği (klavikula) ve çene
bulunur. Etkilenen bölgede ağrı ve şişme oluşabilir.
Bazı durumlarda,
kendiliğinden veya düşme gibi küçük travmayı takiben bir kırık oluşana kadar
hiçbir belirti görülmez. Akut bir lokalize ağrı ve şişme oluşabilir. Daha
yaygın olarak, belirgin bir nedeni olmayan ağrı, zaman içinde frekans ve
yoğunlukta artar ve sonunda alanın zayıflığı ve belirgin deformitesi ile
birlikte olabilir. İlerleme oranı tahmin edilemez ve prognoz zor olabilir.
Hastalık birkaç yıl sonra stabilize olabilir, spontan remisyona girebilir veya
göğüs ve üst omurga içeren vakalarda ölümcül olabilir. Remisyondan sonra
hastalığın tekrarlaması da ortaya çıkabilir. Omurga ve kafatası tabanının
tutulması nörolojik komplikasyonlardan kötü bir sonuca neden olabilir. Birçok
durumda, Gorham hastalığının sonucu ciddi deformite ve fonksiyonel sakatlıktır.
Hastalık kaburgalarda,
skapulada veya torasik omurlarda mevcutsa nefes almada zorluk ve göğüs ağrısı
gibi belirtiler görülebilir. Bunlar, hastalığın kemikten göğüs boşluğuna
yayıldığını gösterebilir. Solunum problemleri astım olarak yanlış teşhis
edilebilir, çünkü akciğerlere verilen hasar, astımda görülen akciğer fonksiyon
testinde aynı tür değişikliklere neden olabilir. Lezyonların göğse uzatılması,
şilöz plevral ve perikardiyal efüzyonların gelişmesine yol açabilir. Chyle,
enfeksiyonla mücadelede önemli olan protein ve beyaz kan hücreleri bakımından
zengindir. Chyle’nin göğse kaybolması, enfeksiyon, yetersiz beslenme ve solunum
sıkıntısı ve başarısızlığı gibi ciddi sonuçlara neden olabilir. Bu
komplikasyonlar veya solunum güçlüğü, göğüs ağrısı, zayıf büyüme veya kilo
kaybı ve enfeksiyon gibi semptomları bazen durumun ilk belirtileri olmuştur.
Gorham hastalığının spesifik
nedeni bilinmemektedir. 1990’lı yıllardan başlayarak, hastalığı olan kişilerde
interlökin-6 (IL-6) adı verilen bir proteinin yüksek seviyelerinin tespit
edildiği bildirildi ve bu da bazılarının artmış IL-6 ve vasküler endotelyal
büyüme faktörünün (VEGF) seviyelerinin katkıda bulunabileceğini düşündürdü.
1999 yılında Möller ve
arkadaşları, “Gorham-Stout sendromu, esasen, artan sayıda parakrin veya otokrin
ile uyarılan hiperaktif osteoklast nedeniyle ciddi bir şekilde artmış kemik
rezorpsiyonu olan monosentrik bir kemik hastalığı olabilir. osteoklastların
varlığı veya yokluğu veya osteoklast sayısı ile ilgili belirgin çelişki,
sendromun farklı evreleri ile açıklanabilir. ” Ayrıca histopatolojik
çalışmalarının Gorham hastalığında görülen osteolitik değişikliklerin
hiperaktif osteoklastik kemiğin sonucu olduğuna dair iyi kanıt sunduğunu
belirtmişlerdir. Bununla birlikte, diğerleri lenfanjiyotoz ve Gorham
hastalığının ayrı hastalıklardan ziyade bir hastalık spektrumu olarak
değerlendirilmesi gerektiği sonucuna varmıştır.
Gorham’ın dengesiz
osteoklastik aktiviteden kaynaklandığına dair bir fikir birliği olsa da, bu
davranışın başlamasına neden olan şey hakkında kesin bir kanıt bulunamamıştır.
Kalıtım Paterni/Deseni
Bugüne kadar literatürde
yaklaşık 300 vaka bildirilmiştir. GSD açık bir ırk, cinsiyet tercihi (1.6: 1;
erkek: kadın oranı) veya coğrafi dağılım göstermemektedir. GSD’nin kesin nedeni
bilinmemektedir. GSD çok nadir
olduğu için, birçok vaka teşhis edilmez veya yanlış teşhis edilir, bu da
bozukluğun genel popülasyondaki gerçek sıklığını belirlemeyi zorlaştırır.
Teşhis ve Tedavi Yöntemleri
Teşhis
Tanı progresif osteolizi ve
kortikal yıkımı gösteren radyografik bulgulara dayanır. Manyetik rezonans
görüntüleme, kemikte tam rezorpsiyon ve T1 ağırlıklı görüntülemede düşük sinyal
yoğunluğu ve T2’de yüksek sinyal yoğunluğu olan ve kontrast görüntülemede yoğun
artış gösteren infiltratif yumuşak doku ile yer değiştirmeyi gösterir. Lenfatik
endotelyal hücrelerin (LYVE-1, podoplanin / D2-40) immünohistokimyasal
belirteçleri, kemiklerin medüller ve kortikal bölgelerinde ve etkilenen yumuşak
dokularda lenfatik damarların varlığını ortaya çıkarır. Kaburga lezyonları
biyopsi yapılmamalıdır, çünkü bu prosedür refrakter bir şilöz efüzyon ortaya
çıkarabilir.
Tedavi
GSD’nin tedavisi, ilerleyici
hastalığı stabilize etmek için ilaçları (bisfosfonatlar ve / veya interferon
alfa 2b, sirolimus da araştırılmaktadır) ve şilotoraksı azaltabilen veya
durdurabilen destekleyici prosedürleri (plörektomi, plörodez, torasentez ve
torasik kanal embolizasyonu veya ligasyonu) içerebilir veya iskeletin etkilenen
bölgelerini stabilize edebilir. Radyoterapi bu terapilerle kombinasyon halinde
kullanılabilir, ancak genellikle refrakter veya hızla ilerleyen hastalıklara
ayrılır.
Prognoz, etkilenen alanların
boyutuna ve yerine bağlıdır. Hafif hastalık yıllarca stabil kalabilirken,
kraniyofasiyal ve / veya torasik bölgeleri içeren ciddi vakalar ölümcül
olabilir. Akciğer tutulumu kötüleşen bir prognozun göstergesidir.
Gorham hastalığının tedavisi
çoğunlukla palyatiftir ve semptom yönetimi ile sınırlıdır.
Bazen kemik yıkımı
kendiliğinden sona erer ve tedavi gerekmez, ancak hastalık ilerlediğinde
agresif müdahale gerekebilir. Duffy ve meslektaşları, kaburga, omuz veya üst
omurgada Gorham hastalığı olan kişilerin yaklaşık% 17’sinin, hastalığın göğsüne
genişlemesini yaşadığını ve ciddi sonuçlarıyla şilotoraksa yol açtığını ve bu
gruptaki ölüm oranının cerrahi müdahale olmadan% 64’e kadar çıkabilir.
Şilomikron Retansiyon Hastalığı besinlerden yağ, kolesterol
ve bazı vitaminlerin alımının bozulduğu kalıtsal bir hastalıktır. Başlıca
sindirim sistemini ve sinir sistemini etkiler. Bu hastalık bebeklik ya da erken
çocukluk çağında ortaya çıkar. Etkilenen çocuklarda kandaki kolesterol seviyesi
düşüktür. Ayrıca E vitamini eksikliği, büyüme geriliği, sürekli ishal, kötü
kokulu dışkı görülür.
Belirti ve
Semptomlar
Hastaların tümünde kolesterol düşüklüğü ve ishal görülür.
Bunların yanında bireylere göre çeşitlilik gösteren birçok belirti vardır.
Yüksek seviyede karaciğer enzimleri, retina hastalıkları, dışkıda yağ fazlalığı
(steatore), şişkinlik, vitamin metabolizması bozuklukları da etkilenen
bireylerde görülen belirtilerdendir. Çocukluk çağının sonlarında sinir
sisteminde bozukluklar da gelişebilir.
Görülme
Sıklığı
Dünya çapında bugüne kadar yaklaşık 55 vaka tanımlanmıştır.
Genetik
Değişiklikler
SAR1B genindeki
mutasyonlar Şilomikron Retansiyon Hastalığına neden olmaktadır. SAR1B geni Sar1b proteinini kodlar. Bu
protein şilomikron denen moleküllerin taşınmasında rol oynar. Şilomikronlar
sindirim sırasında besin öğelerinin emilimini sağlayan ve yağların, yağda
çözünen vitaminlerin ve kolesterolün ince bağırsaktan kana taşınmasını sağlayan
moleküllerdir. SAR1B geninde meydana
gelen mutasyonlar sonucu bu besin maddelerinin emilimi ciddi ölçüde
azalmaktadır.
Kalıtım
Paterni
Bu hastalık otozomal resesif (çekinik) kalıtılır. Yani
hastalığın oluşması için her iki gen kopyasında da mutasyon olması gerekir.
Teşhis
Yöntemleri
Genelde spesifik semptomlar olmadığı için hastalığın teşhisi
zordur. Teşhis, hastada görülen kronik ishal, yağ emilim bozukluğu ve anormal lipit
profiline (normal trigliserit varlığında, yaklaşık %50 düşük kolesterol) göre
konulabilir. Genetik tanı testleri ile SAR1B
genindeki mutasyonun belirlenmesi mümkündür.
Tedavi
Yöntemleri
Tedavi yaklaşımı beslenme ve büyümenin takibine yönelik
olmalıdır. Uygulanacak tedavi, semptomların belirlenmesi ve önlenmesine
odaklanır. E vitamini eksikliğinin kontrolü nörolojik problemlerin ortaya
çıkmaması için ciddi derecede öneme sahiptir. Tedavi, yağda çözünen vitaminler
ve büyük miktarda E vitamini takviyesi içerir. Diğer belirtilerin önlenmesi
için de yağ alımının çok sıkı takibini içeren özel diyetler uygulanmaktadır.
Hastalığın
Diğer İsimleri
CMRD, Anderson Sendromu, Bağırsakta yağ emilim bozukluğu,
Bağırsak hücrelerinde Hipobetalipoproteinemi’yle birlikte apolipoprotein b
benzeri protein birikimi





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}