Büşra DURAK
Genel Bilgi
Kalıtsal evrensel diskromatozis (DUH), bebeklik veya erken çocukluk döneminde ortaya çıkan; gövde, uzuvlar ve bazen yüz üzerinde de genel bir dağılımla ortaya çıkan düzensiz şekilli, asemptomatik hiper ve hipopigmente maküllerle karakterize nadir görülen bir cilt pigmentasyon hastalığıdır.
Genetik Değişiklikler/Etken Faktörler
DUH’nin etiyolojisi hala belirsizdir, ancak histopatoloji tipik olarak bazal tabakanın melanin içeriğinde (biyopsi yapılan lezyonun tipine bağlı olarak) fokal bir artış veya azalma ve bazen de pigmenter inkontinans gösterir. Yapılan bir ultrastrüktürel cilt araştırmasında DUH’nin bir melanosit sayısı bozukluğu değil, melanozom sentez hızı veya melanosit aktivitesi bozukluğu olduğu belirtilmiştir.
Deri pigmenti melanin, epidermisin bazal tabakasında bulunan melanositler tarafından sentezlenir. Melanositlerde melanin sentezi, endoplazmik retikulumdan türetilen melanozomlar içinde gerçekleşir.
Eksozom salgısının melanozom taşıma makineleriyle ilgili olduğu bulunmuştur. Eksozomlar, multiveziküler cisimlerin hücre yüzeyi plazma zarı ile füzyonu üzerine salgılanan multiveziküler cisimlerde bulunan veziküllerdir. ABCB6 geninin eksozomlarda mevcut olduğu bildirilmiştir. Bu yüzden mutant bir ABCB6 geni, melanozom taşınmasını bozabilir ve DUH’ye neden olabilir.
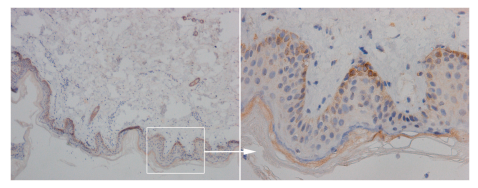
Resim 1. Epidermisin bazal katmanlarının görünümü
(Resim 1 Kaynak: https://www.jidonline.org/article/S0022-202X(15)36415-0/fulltext )
Belirti ve Semptomlar
Klinik olarak gövde ve uzuvlarda değişen boyutlarda çok sayıda hiperpigmente ve hipopigmente makül ile karakterizedir. Etkilenen bireylerin çoğunda lezyonlar 6 yaşına kadar ortaya çıkar, ancak geç başlangıçlı tezahür de bildirilmiştir. Yüz lezyonları, etkilenen bireylerin %50’sine kadar görülür ve avuç içi ve ayak tabanlarının tutulumu olağan dışıdır. Nadiren saç, tırnak ve mukozayı tutabilir. Bazı vakalara sağırlık, görme bozukluğu ve nörolojik semptomlar gibi sistemik hasarlar da eşlik edebilir.
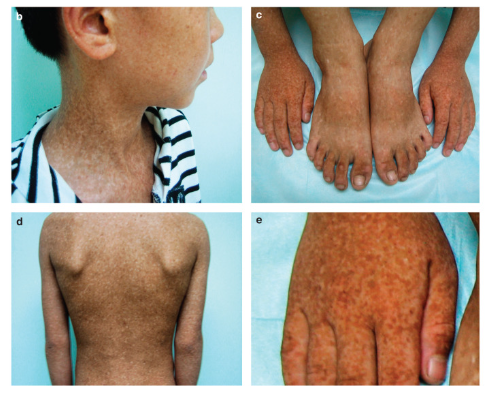
Resim 2. Klinik Özellikler
(Resim 2 Kaynak: https://www.jidonline.org/article/S0022-202X(15)36415-0/fulltext )
Genetik Görülme Sıklığı
Dünya çapında birçok farklı popülasyonda vakalar tanımlanmış olmasına rağmen, ağırlıklı olarak Japon kökenli bireylerde görülür.
Bir çalışma, Japonya’da 100.000 dermatoloji konsültasyonu başına yaklaşık 0.3’ün DUH ile ilgili olduğunu bildirmiştir.
Kalıtım Paterni/Deseni
Vakaların çoğu otozomal dominant bir şekilde kalıtılır ancak otozomal resesif ve sporadik vakalar da bildirilmiştir.
Kalıtsal evrensel diskromatozis (DUH); sırasıyla 6q24.2-q25.2, 12q21q23 ve 2q35 kromozomlarında bulunan farklı bağlantı bölgelerine dayalı olarak, DUH1 (Online Mendelian Inheritance in Man (OMIM) 127500), DUH2 (OMIM 612715) ve DUH3 (OMIM 615402) olmak üzere üç tipe ayrılabilir. DUH1 ve DUH3 otozomal dominant bir şekilde kalıtılırken, DUH2 otozomal resesif bir şekilde kalıtılır.
Teşhis Yöntemleri ve Tedaviler
DUH; genellikle hasta öyküsü, fizik muayene ve cilt biyopsisi ile teşhis edilebilir. Işığa maruziyet sonucunda hastalığın seyrinin değişmemesi, atrofi ve telenjiektazinin nadir olması, lezyonların iyi huylu seyir göstermesi gibi sahip olduğu özelliklerden dolayı bazı cilt hastalıklarından ayırt edilebilir.
Şu anda, DUH’de cilt pigmenter değişiklikleri için etkili bir tedavi yoktur, ancak Q-switched alexandrite lazer tedavisi, hiperpigmente lezyonlar için özellikle açıkta kalan alanlarda (örneğin: yüz ve eller) kullanılabilir.
Hastalıkla İlişkili Genler
ABCB6 ve SASH1, DUH ile ilgili yakın zamanda bildirilen patojenik genlerdir.
ABCB6 geni vücudun farklı bölgelerinde geniş bir fonksiyona sahiptir; bu nedenle, ABCB6 alanlarının mutasyonu, çeşitli fenotiplerde hastalık üretebilir. Ancak, tüm DUH hastaları ABCB6 mutasyonuna sahip değildir. DUH’nin genetik heterojen bir hastalık olması mümkündür.
Hastalığın Diğer İsimleri
Yok.
Kaynakça
- https://www.jaadcasereports.org/article/S2352-5126(21)00865-1/fulltext
- https://www.jidonline.org/article/S0022-202X(15)36415-0/fulltext
- https://www.ejdv.eg.net/article.asp?issn=1110-6530;year=2016;volume=36;issue=1;spage=26;epage=27;aulast=Gupta
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3911924/
- https://assets.cureus.com/uploads/case_report/pdf/65233/20210819-29838-tu8d6b.pdf
- https://journals.lww.com/idoj/pages/articleviewer.aspx?year=2020&issue=11020&article=00022&type=Fulltext
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8236144/#CR1
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3911924/
- https://escholarship.org/uc/item/29m212t0
- https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=8630&Disease_Disease_Search_diseaseGroup=-Dyschromatosis-Universalis-Hereditaria&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Dyschromatosis-universalis-hereditaria&title=Dyschromatosis%20universalis%20hereditaria&search=Disease_Search_Simple
- https://www.visualdx.com/visualdx/diagnosis/dyschromatosis+universalis+hereditaria?diagnosisId=56260&moduleId=101
- https://rarediseases.info.nih.gov/diseases/1996/dyschromatosis-universalis-hereditaria/research
- http://www.odermatol.com/odermatology/2020e/E189.dyschromatosis-BasavarajNY.pdf
- https://www.omim.org/entry/127500?search=Dyschromatosis%20universalis%20hereditaria&highlight=dyschromatosi%20hereditaria%20universali
- https://www.uptodate.com/contents/the-dyschromatoses#H1469243