Hilal Altay
Genel Bilgi, Genetik Değişiklikler/Etken Faktörler
Birincil Sklerozan Kolanjit(PSC) yaklaşık olarak 40’lı yaşlarda teşhis edilmekle beraber, bilinmeyen nedenlerden erkeklerde kadınlardan iki kat fazla sıklıkta görülmektedir.
- Belirti ve Semptomlar
Teşhis edilen hastaların çoğunda hastalığın hiçbir semptomu görülmemekte, fakat rutin kan testleri karaciğerdeki sorunları ortaya çıkarmaktadır. En erken görülen PSC semptomu aşırı yorgunluk (fatigue), karın bölgesinde rahatsızlık ve aşırı kaşıntıdır(pruritus). Durum kötüleştikçe, hastalıktan mustarip bireyler deride ve göz beyazında sarılık ve/veya dalakta büyüme(splenomegaly) gösterebilir. Biriken safra son aşamalarda karaciğere büyük zarar vererek kronik karaciğer hastalıklarına(siroz) ya da karaciğer yetmezliğine neden olur. Sindirmek için gerekli safraya erişilemediğinden, yağın vücutta parçalanması/emilimi gerçekleşemez. Bunun sonucunda, kilo kaybı ve yağ içerisinde bulunan ya da yağ içerisinde depolanan (yağda çözünen) vitaminlerin eksikliği ortaya çıkar. Örneğin yağda çözünen D vitamini kalsiyumu absorbe etmek ve bu doğrultuda kemikleri güçlendirmekten sorumludur. Bu vitaminin eksikliği PSC’ye sahip bireylerde kemik incelmesinin (osteoporoz) görülmesine neden olmaktadır.
- Genetik Görülme Sıklığı
Her 10,000 insanda 1 görülen bu hastalık, yılda yaklaşık olarak her 100,000 hastanın birinde teşhis edilebilmektedir.
- Kalıtım Paterni/Deseni
Hastalığın genetik paterni, hastalığın çeşitli ve çok sayıdaki genetik ve çevresel faktörlerin etkileşimi içerisinde olmasından dolayı tam olarak bilinmemektedir. Bu hastalık aile içerisinde kümelenme eğiliminde olduğundan, herhangi bir aile bireyinin bu hastalığa sahip olması diğer aile üyelerinin de hastalığa sahip olma açısından riskli konumda olduğunu göstermektedir.
- Teşhis Yöntemleri ve Tedaviler
PSC, klinik belirtilere ve değişen karaciğer fonksiyon testlerine dayanılarak hastalıktan şüphelendiğinde, hastalığın karakteristiği olan safra kanalında darlık ve dilatasyon değişikliklerin görüntülenmesi amacıyla yapılan manyetik rezonans kolanjiyopankreatografi (MRCP) veya endoskopik retrograd kolanjiyopankreatografi (ERCP) ile tanı .
İmmünsüpresanlar ve şelatörler semptomları tedavi etmek ve komplikasyonları yönetmek için kullanılır, ancak bugüne kadar PSC’nin hastalık seyrini iyileştiren veya değiştiren hiçbir tıbbi tedavi bulunmamaktadır. Yağda çözünen A, D, E ve K vitaminlerinin yerine koyma tedavisi hastalarda sıklıkla görülen vitaminlerin eksikliğini gidermek için kullanılabilir. Karaciğer nakli hastalığı ileri seviyede olan hastalar için tek kesin tedavi yöntemidir ve hastaların çoğunda tanıdan 13-21 yıl sonra karaciğer nakli, ya hastalığın ilerlemesi ya da tedaviye dirençli bakteriyel kolanjit gibi hastalık komplikasyonları nedeniyle gerekmektedir. Safra tıkanıklığı semptomlarını hafifletmek için, safra darlıkların stentli veya stentsiz endoskopik dilatasyonu .
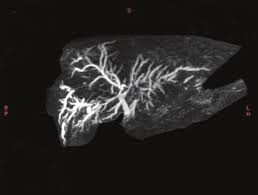
Karaciğer nakli sonrası hastalığın seyri genellikle iyidir, ancak karaciğer nakli gerçekleştirilen PSC hastalarının % 10 ila %37’lik bir kısmında hastalık tekrarlayabilmektedir.
- Hastalıkla İlişkilendirilen Diğer Hastalıklar
PSC genellikle bağırsakların iltihaplanmasıyla bağırsaklarda açık yaralara(ülser) ve karın ağrısına neden olan iltihaplı bağırsak hastalığı (IBD) ile ilişkilendirilmektedir. Ama bu bağlantının nedeni hala anlaşılamamıştır. PSC hastalığından mustarip kişilerin yaklaşık %70’i IBD’nin en yaygın formu olan ülseratif kolit’e sahiptir. Bunun yanı sıra, PSC hastalığından mustarip bireylerin Tip 1 Diyabet, Çölyak ve Tiroid hastalığı gibi otoimmün hastalıklarına yakalanma şanları hastalığa sahip olmayan bireylerden daha fazladır. Otoimmün hastalıkları, bağışıklık sisteminin arızalanması sonucu vücut organ ve dokularına saldırması ile gerçekleşmektedir. PSC hastalığından mustarip kişilerin kansere, özellikle safra kanalı kanseri (Kolanjiokarsinoma), yakalanma riskleri de artmaktadır.
- Hastalığın Diğer İsimleri
- PSC
- Sclerosing Cholangitis
- Hastalık adına Yapılan Çalışmalar ve Kısa Özetleri
Flow Cytometry kullanarak Eksteen vd. organ donör kontrolleri ve diğer kronik inflamatuar karaciğer hastalıkları olan hastalara kıyasla primer sklerozan kolanjiti olan hastalarda karaciğer infiltre eden lenfositler (LIL’ler) üzerinde CCR9’un (604738) büyük ölçüde artmış ekspresyonunu bulmuştur. İmmünohistokimyasal ve Western Blot analizleri, normalde sadece timüs ve bağırsakta eksprese edilen CCL25’in (602565), PSC hastalarının karaciğerlerinde yüksek oranda eksprese edildiğini, ancak diğer karaciğer hastalıklarında böyle bir olayın gerçekleşmediği belirtildi. Laboratuvar ortamında CCR9-pozitif LIL’ler seçilerek CCL25’e taşınmıştır ve CCL25’lerin alfa-4 / beta-7 integrinleri aracılığıyla hareket halinde bulunmayan MADCAM1’e (102670) için tetiklenebileceği gözlenmiştir5. Eksteen vd., aktif enflamatuar bağırsak hastalığı sırasında bağırsakta aktive olan T hücrelerinin hem hepatik hem de mukozal endotelyuma bağlanma yeteneği ile efektör hücrelere farklılaştığını öne sürmüştür. Eksteen vd. bu çalışmalarında CCL25 ve MADCAM1’in PSC’de karaciğere mukozal CCR9-pozitif lenfositlerin alınmasında beraber çalıştıklarını öne sürmüştür.
Sheth vd., CFTR’nin (602421) işlev bozukluğunun, IBD’li bir hasta alt kümesinin neden PSC geliştirdiğini açıklayabileceğini varsaydı. PSC’li 19 hastada CFTR genotipini ve fenotipini IBD’si olan ve karaciğer hastalığı olmayan 18 hasta, 17 primer biliyer siroz hastası, 81 kistik fibroz hastası(CF; 219700) ve 51 sağlıklı kontrol ileriye dönük olarak değerlendirdiler. Moleküler ve fonksiyonel analizlerle gösterildiği üzere, PSC’de heterozigot durumda CFTR anormalliklerinin prevalansının arttığını buldular ve bu anormalliklerin IBD’li bir hasta alt kümesinde PSC’nin gelişimine katkıda bulunabileceği sonucuna vardılar. PSC hastalarının yüzde seksen dokuzu, 1540G varyantını (602421.0023) içeren genotipleri taşımakla beraber hastalık kontrollerinin% 57’sine kıyasla (p = 0.03) azalmış fonksiyonlu CFTR’a sahip oldukları anlaşıldı. 19 PSC hastasından sadece 1’inde ne CFTR mutasyonu ne de 1540G varyantı vardı. Nazal potansiyel fark testi ile değerlendirilen CFTR klorür kanal fonksiyonu, hastalık kontrolleri ve sağlıklı kontroller ile karşılaştırıldığında PSC hastalarında medyan izoproterenol yanıtının azaldığını göstermiştir.
Takeda vd., bir monoklonal anti Dr (603612) antikoruuygulanmasının, B farelerinde spesifik olarak kolestatik karaciğer yetmezliği ve kolanjiyosit apoptozunu indüklediğini, ancak diğer fare suşlarında olmadığını bulmuştur. Trail (603598) – veya Dr-null B fareleri, genel safra kanalı ligasyonu ile indüklenen kolestaza nispeten dirençliydi7. Ek olarak, ortak safra kanalı ligasyonu, yabani tip farelerin kolanjiyositlerinde Dr ekspresyonunu artırarak onları Dr aracılı kolanjite duyarlı hale getirdi. Fare karaciğer örneklerinin histolojik görünümü, insan primer sklerozan kolanjitini . PSC’li hastalardan türetilen kolanjiyositler, kontrollere kıyasla DR5 ve TRAIL ekspresyonunda artış . İnsan örneklerinde kolanjiyositlerin apoptozu da gözlendi. Bulgular, sirotik safra hastalıklarında TRIAL / DR5 aracılı apoptoz için bir rol önermiştir.
Hastalık ile ilgili devam eden veya tamamlanmış laboratuvar deney çalışmalarına bu siteden ulaşılabilmektedir:
https://clinicaltrials.gov/ct2/resultscond=%22primary+sclerosing+cholangitis%22
KAYNAKÇA
- Primary sclerosing Cholangitis: MedlinePlus Genetics. (2020, August 18). https://medlineplus.gov/genetics/condition/primary-sclerosing-cholangitis/#causes adresinden alıntılandı.
- INSERM US14 — ALL RIGHTS RESERVED. (n.d.). Orphanet: Primary sclerosing cholangitis. https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=783&Disease_Disease_Search_diseaseGroup=Primary-sclerosing-cholangitis-&Disease_Disease_Search_diseaseType=Pat&Disease%28s%29%2Fgroup+of+diseases=Primary-sclerosing-cholangitis&title=Primary+sclerosing+cholangitis&search=Disease_Search_Simple adresinden alıntılandı.
- Primary Sclerosing Cholangitis. (n.d.). https://omim.org/entry/613806#1 adresinden alıntılandı.
- Primary sclerosing cholangitis. (2019, September 12). https://rarediseases.org/rare-diseases/primary-sclerosing-cholangitis/ adresinden alıntılandı.
- Eksteen, B., Grant, A. J., Miles, A., Curbishley, S. M., Lalor, P. F., Hubscher, S. G., Briskin, M., Salmon, M., Adams, D. H. Hepatic endothelial CCL25 mediates the recruitment of CCR9+ gut-homing lymphocytes to the liver in primary sclerosing cholangitis. J. Exp. Med. 200: 1511-1517, 2004.
- Sheth, S., Shea, J. C., Bishop, M. D., Chopra, S., Regan, M. M., Malmberg, E., Walker, C., Ricci, R., Tsui, L.-C., Durie, P. R., Zielenski, J., Freedman, S. D. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum. Genet. 113: 286-292, 2003.
- Takeda, K., Kojima, Y., Ikejima, K., Harada, K., Yamashina, S., Okumura, K., Aoyama, T., Frese, S., Ikeda, H., Haynes, N. M., Cretney, E., Yagita, H., Sueyoshi, N., Sato, N., Nakanuma, Y., Smyth, M. J., Okumura, K. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc. Nat. Acad. Sci. 105: 10895-10900, 2008.