Genel Bilgi
Kondroektodermal displazi , ekstremitelerin normalden kısa oluşu , fazladan el/ayak parmakları (polidaktili) , anormal tırnak gelişimi ve olguların yarısından fazlasında konjenital kalp defektleriyle karakterize nadir görülen bir genetik hastalıktır . Motor gelişim ve zeka normaldir.

Görsel 1 : Polidaktili
Kaynak : https://ghr.nlm.nih.gov/art/large/polydactyly.jpeg

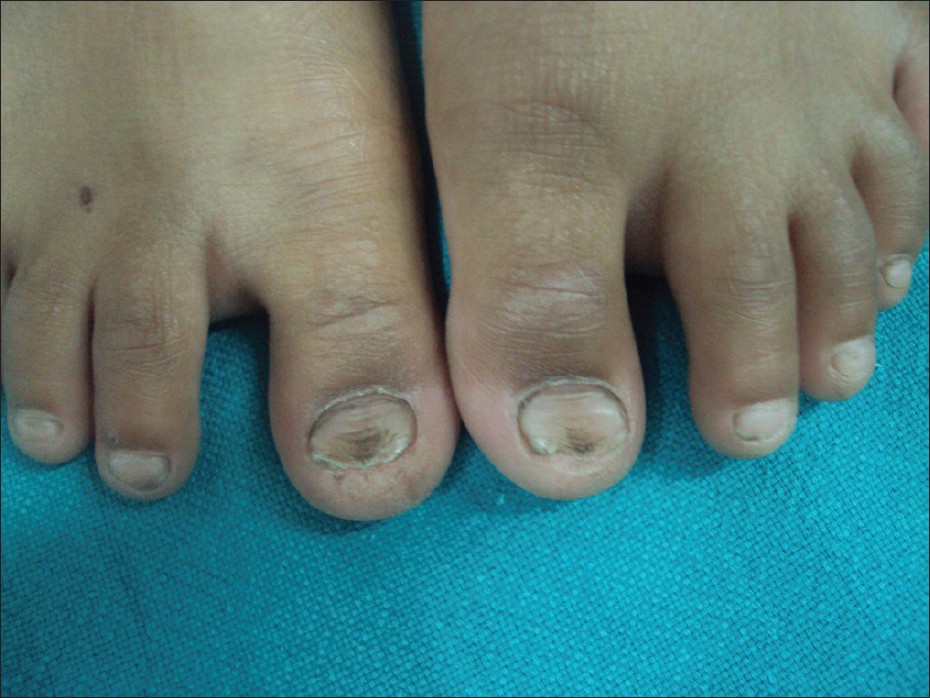
Görsel 2 : Hipoplastik tırnaklar
Kaynak : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3354796/figure/F1/
Hastalığın diğer bilinen isimleri
- Kondroektodermal displazi
- Ellis-Van Creveld displazisi
Genetik Değişiklikler
Kondroektodermal displazi EVC veya EVC2 genlerindeki mutasyonlar sonucu oluşabilir . Bu genlerin fonksiyonu hakkında çok az şey bilinmesine rağmen gelişim esnasında hücreden-hücreye sinyal iletiminde önemli görevleri olduğu görülmektedir . Ayrıca , EVC ve EVC2 genlerinden üretilen proteinlerin Sonic Hedgehog sinyal yolağında düzenleyici göreve sahip olduğu düşünülmektedir . Bu yolak hücre büyümesinde , özelleşmesinde ve vücudun çoğu bölgesinin normal şekillenmesinde önemli bir rol oynamaktadır .
Kondroektodermal displazi’ne sebep olan mutasyonlar , anormal şekilde küçük , işlevsiz EVC/EVC2 proteinlerinin üretimine sebep olurlar . Hatalı proteinlerin bu duruma nasıl sebebiyet verdiği tam olarak anlaşılamamıştır . Çalışmalar bu hatalı proteinlerin Sonic Hedgehog sinyal yolağının normal işleyişini engellediğini böylelikle embriyo gelişimi sürecinde kemiklerin , dişlerin ve vücudun diğer bölümlerinin oluşumunu ve büyümesini bozduğunu göstermektedir .
EVC ve EVC2 genlerinde mutasyon görülmesi Kondroektodermal displazi
yarısından fazlasında görülen bir durumdur . Geri kalan olguların sebebiyse henüz bilinmemektedir.
Belirti ve Semptomlar
Kondroektodermal displazi bireyler , tipik olarak kısa bacak ve kollara sahiptirler . Bunun yanı sıra baş ve gövdeleri normaldir . Bu hastalığa sahip tüm bireylerde ekstra parmaklar(polidaktili) görülmektedir ve her iki el de genellikle etkilenmiştir . Ektodermal anormaliler , örneğin saç tırnak ve dişlerin anormal gelişimi de görülmektedir .
Kondroektodermal displazi’li bireylerin yarısından fazlası kalp defektleriyle doğar . En yaygın kalp defektiyse atrial septal defekttir . Ventriküler septal defekt ve patent ductus arteriosus gibi diğer kalp defektleri de görülebilir .
Bu sendroma sahip erkeklerin bazılarında inmemiş testis(kriptorşidizm) veya idrar yolu çıkışının, normalde olduğu gibi penisin ucuna değil, üst yüzeyine açılmasıyla beliren doğumsal oluşum bozukluğu(epispadias) sahip olduğu görüşmüştür . Göğüs duvarında , omurgada ve solunum sisteminde anomaliler de belirtilmiştir .


Görsel 3 : Kriptorşidizm
Kaynak : http://www.health-tutor.com/cryptorchidism.html


Görsel 4 : Epispadias
Kaynak : https://www.epainassist.com/pelvic-pain/urethra/epispadias
Görülme Sıklığı
Dünyanın çoğu bölgesinde , Ellis- Van Creveld sendromu 60.000 – 200.000 yenidoğanda 1 görülür . Hastalık çok nadir gözüktüğünden kesin prevalansı saptamak çok güçtür . Sendrom özellikle Lancaster vilayetinde Old Order Amish popülasyonunda , Pensilvanya’da , ve Batı Avustralya’ın yerli popülasyonunda daha sık görülmektedir.
Kalıtım Paterni
Bu sendrom otozomal resesif olarak kalıtılır bu da demek oluyor ki her hücredeki iki kopyada da mutasyon bulunmaktadır .Otozomal resesif bir hastalığın ebeveynleri mutasyona uğramış genlerin birer kopyasını taşımaktadır ancak hastalığın tipik bulgu ve belirtilerini göstermezler.
Teşhis
Ellis-Van Creveld sendromu , kısa boy ve yavaş büyümenin gözlemlenmesi , görüntüleme teknikleriyle belirlenmiş iskelet anomalileri veya bazen doğuştan diş(natal diş) bulunmasıyla tanı alabilir . EVC ve EVC2 genleri için moleküler genetik çalışmalar sadece araştırma amaçlı kullanılmaktadır . Ultrasonla prenatal tanı mümkündür.
Tedavi
Sıklıkla ,doğumdan kısa süre sonra göğüs darlığı ve/veya kalp yetmezliği sonucu gelişen solunum sıkıntısı tedavi edilmelidir. Natal dişler çıkarılmalıdır çünkü beslenmeyi sekteye uğratabilir.
Ellis-Van Creveld sendromunun tedavisi , her bireyde gözüken semptomlara spesifik yapılmalı . Bu tarz bir tedavi , pediatri uzmanları , cerrahlar , kardiyoloji uzmanları , diş doktorları , göğüs hastalıkları , ortopedi , üroloji uzmanları ve terapistlerin birlikte koordineli çalışmasıyla mümkündür .
Etkilenen bireyler ve aileleri için genetik danışmanlık önerilmektedir .
KULLANILAN KAYNAKLAR
https://rarediseases.org/rare-diseases/ellis-van-creveld-syndrome/
https://ghr.nlm.nih.gov/condition/ellis-van-creveld-syndrome#
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3354796/figure/F1/





{kind=link}